Using Web-Based Computations in Organic Chemistry
|
|
|
- Ashlynn Hodges
- 5 years ago
- Views:
Transcription
1 10/30/ Using Web-Based Computations in Organic Chemistry John Keller UAF Department of Chemistry & Biochemistry The UAF WebMO site Practical aspects of computational chemistry theory and nomenclature MOPAC2016: fast and approximate NWChem: slow and accurate Example tasks: Energy, Optimization, vibrational spectra; UV spectra.


2 10/30/ Click here to log in (guest/webmo or yourusername/password)

3 10/30/ How does a WebMO job work? webserver world wide web User Draw molecule & choose options WebMO creates input file for MOPAC or NWChem When the job is done, the output file is parsed and shown on Results page. The updated, intermediate results can be visualized in the Job Manager. compute servers obsidian antec12 (& others). Running Gaussian, MOPAC, and NWChem
4 10/30/ User Accounts MOPAC NWChem Gaussian CPU job time limit (h) guest 4 uaaguest 12 Non-UAF unlimited UAF unlimited Machines Server Cores Procs Mem (GB) CPU Gaussian 09 NWChem 6.6 Chemlinux Xeon (2) Chemlinux Xeon (2) Chemlinux Xeon (2) Antec Core i7 5820K Corsair Core i7 6950X Obsidian Core i7 930 MOPAC 2016
5 10/30/ Levels of theory Correlated ab initio MO (MP2 & others) Molecular mechanics (MM+, OPLS, etc) Ball-andspring model no orbitals no electronics Semi-empirical MO (PM3, PM6, PM7) Uses 27 orbital parameters for each element Parameters derived from stable molecules Electron correlation implicit, since the parameters are fr real molecules, where actual electron correlation occurs. Ab initio MO (Hartree-Fock HF) MOs = linear combination of basis orbitals. Parameters derived from isolated atoms Some electron correlation Density Functional Theory (DFT) (B3LYP ) MOs = linear combination of basis orbitals Parameters derived from atoms and molecular databases More electron correlation MOs = linear combination of basis orbitals. Parameters derived from isolated atoms Includes excited state configurations Most electron correlation
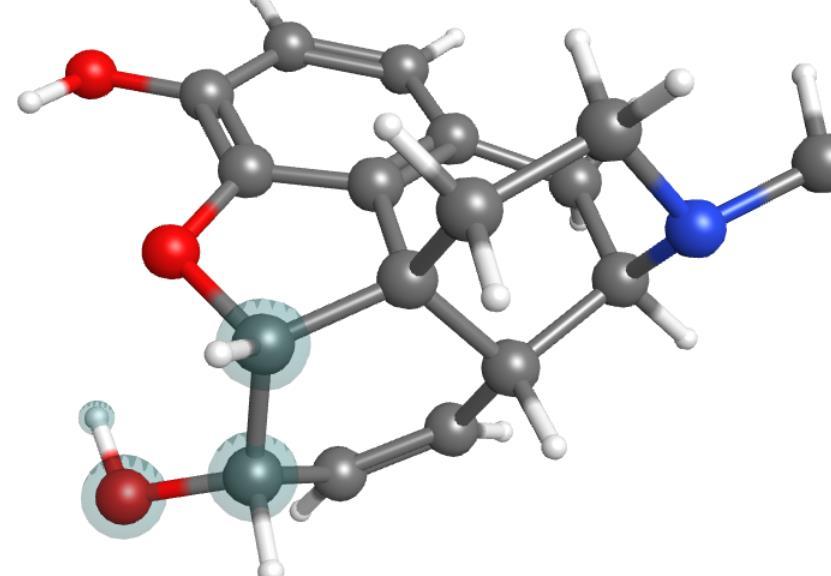
6 10/30/ Basic WebMO and MOPAC 1) Build hexatriene; load and modify morphine. 2) Optimize Use MOPAC PM7
7 10/30/ Log in

8 10/30/ Begin with New Job, Create New Job
9 10/30/2017 9


10 10/30/ Select with mouse. Then use the Adjust menu to change torsion angles, bond distances
 Engine: MOPAC 3) Select Server: First")
11 10/30/ Optimize your molecule 1) Right Arrow 2) Engine: MOPAC 3) Select Server: First Available
 Charge: (0 for molecule) Multiplicity: generally singlet (unless odd # of valence")
12 10/30/ Enter an informative Job Name Type of calculation Geometry Optimization Theory: PM7 (a recent version of PM3 semi-empirical) Charge: (0 for molecule) Multiplicity: generally singlet (unless odd # of valence e - )
13 10/30/ Generate on the Preview tab shows the input file automatically created by WebMO. It can be modified manually. Submit job by clicking the right arrow
14 10/30/
15 10/30/ NWChem software Review MOs Basis sets Performance comparison: system size and basis set choice Typical NWChem jobs: Energy, Opt, Vibrations, UV
16 10/30/ On-line NWChem User s Reference



")


17 H 2 MOs: formed by linear combination of 1s atomic orbitals. 17 Electron density (0.008 e/å 3 ) node node anti-bonding MO H atom H atom bonding MO 1s AO 1s AO d = 0.76 Å d = 2.8 Å d =
,")

 p x, p y, and p")

18 18 In semi-empirical MO theory (PM6 and others), molecular orbitals are formed by linear combinations of s, (and on heavy atoms) p x, p y, and p z AOs. s p x p y p z The orbital functions on the constituent atoms are the basis set.
19 10/30/ NWChem and other QM programs use larger basis sets. The most common are split-valence basis sets. Basis orbital for core electrons of 2 nd row atom contains 6 Gaussian terms. 6-31G(d) 2 nd row atoms have an additional set of six d-type orbitals. Inner valence shell orbital contains 3 Gaussian terms. Outer valence shell orbital contains 1 Gaussian. H 2 O has 19 orbitals in the 6-31G(d) basis. O: 1s + 2s,2p x,2p y,2p z + (2s,2p x,2p y,2p z ) + 2(d xx,d yy,d zz,d xy,d yz,d xz ) H: - 1s + (1s) H: - 1s + (1s)

 Vitamin")
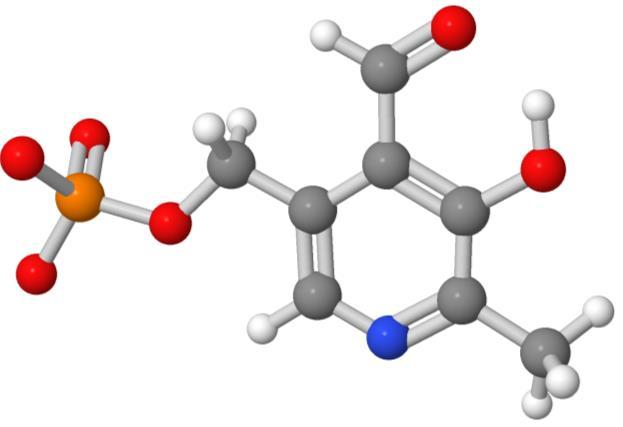
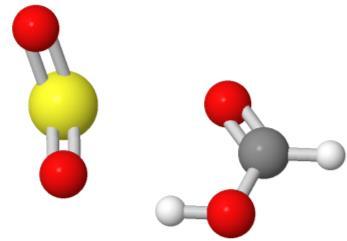
20 10/30/ Tetracycline (32) Vitamin B-6 (16) a-cyclodextrin (66) SO 2 -formic acid (6 heavy atoms)
21 10/30/ # Heavy atoms Basis set/# of basis orbitals 3-21G 6-31G(d,p) G(2d,2p) G(3df,3pd) SO 2 -formic acid Vitamin B Tetracycline a-cyclodextrin
22 10/30/ ~ 20 optimization steps 6.5 da 20 h Basis set Basis set small large Small molecule OK OK Large molecule OK (no) Hartree-Fock; Corsair3; 10 processors
23 10/30/ NWChem input file format Energy Geometry Optimization Vibrational Frequency UV spectrum

24 10/30/ NWChem input file format Energy (single point) Job name charge molecular description Basis set theory multiplicity Calculation type
25 10/30/ NWChem input file format WebMO default - Optimization
26 10/30/ You may wish to add sections to modify some parts of the input file. scf noprint "final vectors analysis" singlet end driver maxiter 100 end NWChem stops after 20 optimization steps by default so, increase the limit this way. task scf optimize


27 10/30/ Geometry Optimization View Results
28 10/30/ NWChem input file format - Vibrational frequencies title "H2O-631Gs-HF-Freq" echo charge 0 geometry O H H end basis noprint * library 6-31G* end scf noprint "final vectors analysis" singlet end task scf freq Sally 3249 pulegone vib

29 10/30/ Vibrational Frequencies View Results


30 10/30/ From Japan Spectral database Pulegone Exp tl B3LYP/6-31G(d) HF/6-31G(d) (This is why one should use HF or DFT methods for IR.)? PM7
31 10/30/ UV-Vis spectra More conjugated double bonds lead to lower energy gap, lower frequency and longer wavelength. DE = hn = hc/l
32 10/30/ NWChem CIS (configuration interaction singles) mixes 10 single excited states Etc

33 10/30/ UV-Vis spectra
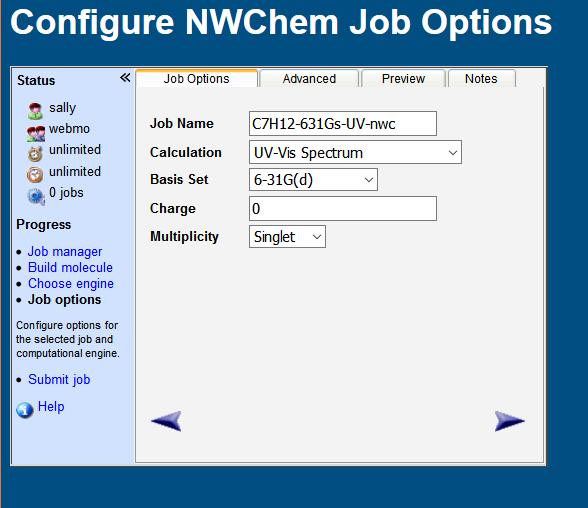
34 10/30/

35 10/30/ C=C yellow Plot line colors are arbitrary. 9 yellow-orange 11 orange-red
36 10/30/ Your questions
Computational Chemistry Using the University of Alaska WebMO Site
 2/7/2017 1 Computational Chemistry Using the University of Alaska WebMO Site John Keller Department of Chemistry & Biochemistry University of Alaska Fairbanks Intro and operation of WebMO and MOPAC Basic
2/7/2017 1 Computational Chemistry Using the University of Alaska WebMO Site John Keller Department of Chemistry & Biochemistry University of Alaska Fairbanks Intro and operation of WebMO and MOPAC Basic
John Keller Department of Chemistry & Biochemistry University of Alaska Fairbanks
 10/15/2016 1 WebMO & Gaussian John Keller Department of Chemistry & Biochemistry University of Alaska Fairbanks Corrections and updates 9-5-2017 SCHEDULE 9-10 Intro and basic operation of WebMO and MOPAC
10/15/2016 1 WebMO & Gaussian John Keller Department of Chemistry & Biochemistry University of Alaska Fairbanks Corrections and updates 9-5-2017 SCHEDULE 9-10 Intro and basic operation of WebMO and MOPAC
Jack Smith. Center for Environmental, Geotechnical and Applied Science. Marshall University
 Jack Smith Center for Environmental, Geotechnical and Applied Science Marshall University -- Division of Science and Research WV Higher Education Policy Commission WVU HPC Summer Institute June 20, 2014
Jack Smith Center for Environmental, Geotechnical and Applied Science Marshall University -- Division of Science and Research WV Higher Education Policy Commission WVU HPC Summer Institute June 20, 2014
IFM Chemistry Computational Chemistry 2010, 7.5 hp LAB2. Computer laboratory exercise 1 (LAB2): Quantum chemical calculations
: Quantum chemical calculations") Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Introductory WebMO Exercises
 Introductory WebMO Exercises These directions assume no prior knowledge of e WebMO interface and provide detailed, click by click instructions on building molecules and setting up calculations. Use your
Introductory WebMO Exercises These directions assume no prior knowledge of e WebMO interface and provide detailed, click by click instructions on building molecules and setting up calculations. Use your
General Chemistry Lab Molecular Modeling
 PURPOSE The objectives of this experiment are PROCEDURE General Chemistry Lab Molecular Modeling To learn how to use molecular modeling software, a commonly used tool in chemical research and industry.
PURPOSE The objectives of this experiment are PROCEDURE General Chemistry Lab Molecular Modeling To learn how to use molecular modeling software, a commonly used tool in chemical research and industry.
Introduction to Hartree-Fock calculations in Spartan
 EE5 in 2008 Hannes Jónsson Introduction to Hartree-Fock calculations in Spartan In this exercise, you will get to use state of the art software for carrying out calculations of wavefunctions for molecues,
EE5 in 2008 Hannes Jónsson Introduction to Hartree-Fock calculations in Spartan In this exercise, you will get to use state of the art software for carrying out calculations of wavefunctions for molecues,
Calculating Bond Enthalpies of the Hydrides
 Proposed Exercise for the General Chemistry Section of the Teaching with Cache Workbook: Calculating Bond Enthalpies of the Hydrides Contributed by James Foresman, Rachel Fogle, and Jeremy Beck, York College
Proposed Exercise for the General Chemistry Section of the Teaching with Cache Workbook: Calculating Bond Enthalpies of the Hydrides Contributed by James Foresman, Rachel Fogle, and Jeremy Beck, York College
Lab #3: Choice of Theoretical Method
 Lab #3: Choice of Theoretical Method These directions assume the user is familiar with the WebMO interface and can build molecules, set up calculations, etc. Exercise 1 - Determine the Proton Affinity
Lab #3: Choice of Theoretical Method These directions assume the user is familiar with the WebMO interface and can build molecules, set up calculations, etc. Exercise 1 - Determine the Proton Affinity
Molecular Orbitals for Ozone
 Molecular Orbitals for Ozone Purpose: In this exercise you will do semi-empirical molecular orbital calculations on ozone with the goal of understanding the molecular orbital print out provided by Spartan
Molecular Orbitals for Ozone Purpose: In this exercise you will do semi-empirical molecular orbital calculations on ozone with the goal of understanding the molecular orbital print out provided by Spartan
Ethene. Introduction. The ethene molecule is planar (i.e. all the six atoms lie in the same plane) and has a high degree of symmetry:
 and has a high degree of symmetry:") FY1006 Innføring i kvantefysikk og TFY4215 Kjemisk fysikk og kvantemekanikk Spring 2012 Chemical Physics Exercise 1 To be delivered by Friday 27.04.12 Introduction Ethene. Ethylene, C 2 H 4, or ethene,
FY1006 Innføring i kvantefysikk og TFY4215 Kjemisk fysikk og kvantemekanikk Spring 2012 Chemical Physics Exercise 1 To be delivered by Friday 27.04.12 Introduction Ethene. Ethylene, C 2 H 4, or ethene,
Gaussian: Basic Tutorial
 Input file: # hf sto-g pop=full Water - Single Point Energy 0 H.0 H.0 H 04.5 Route Section Start with # Contains the keywords Gaussian: Basic Tutorial Route Section Title Section Charge-Multiplicity Molecule
Input file: # hf sto-g pop=full Water - Single Point Energy 0 H.0 H.0 H 04.5 Route Section Start with # Contains the keywords Gaussian: Basic Tutorial Route Section Title Section Charge-Multiplicity Molecule
Lec20 Fri 3mar17
 564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
Introduction to computational chemistry Exercise I: Structure and electronic energy of a small molecule. Vesa Hänninen
 Introduction to computational chemistry Exercise I: Structure and electronic energy of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the electronic energy
Introduction to computational chemistry Exercise I: Structure and electronic energy of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the electronic energy
Density Functional Theory
 Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Exercise 1: Structure and dipole moment of a small molecule
 Introduction to computational chemistry Exercise 1: Structure and dipole moment of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the dipole moment of a small
Introduction to computational chemistry Exercise 1: Structure and dipole moment of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the dipole moment of a small
NH 3 inversion: Potential energy surfaces and transition states CH342L March 28, 2016
 N 3 inversion: Potential energy surfaces and transition states C342L March 28, 2016 Last week, we used the IR spectrum of ammonia to determine the splitting of energy levels due to inversion of the umbrella
N 3 inversion: Potential energy surfaces and transition states C342L March 28, 2016 Last week, we used the IR spectrum of ammonia to determine the splitting of energy levels due to inversion of the umbrella
Lab #5: Electron Densities, Electrostatic Potentials, and Reactivity Indices
 Lab #5: Electron Densities, Electrostatic Potentials, and Reactivity Indices Exercise 1 - Visualizing Different Bond Types Build H 2 and perform a geometry optimization (Mopac) using the choices shown
Lab #5: Electron Densities, Electrostatic Potentials, and Reactivity Indices Exercise 1 - Visualizing Different Bond Types Build H 2 and perform a geometry optimization (Mopac) using the choices shown
NMR and IR spectra & vibrational analysis
 Lab 5: NMR and IR spectra & vibrational analysis A brief theoretical background 1 Some of the available chemical quantum methods for calculating NMR chemical shifts are based on the Hartree-Fock self-consistent
Lab 5: NMR and IR spectra & vibrational analysis A brief theoretical background 1 Some of the available chemical quantum methods for calculating NMR chemical shifts are based on the Hartree-Fock self-consistent
Lec20 Wed 1mar17 update 3mar 10am
 564-17 Lec20 Wed 1mar17 update 3mar 10am Figure 15.2 Shows that increasing the diversity of the basis set lowers The HF-SCF energy considerably, but comes nowhere near the exact experimental energy, regardless
564-17 Lec20 Wed 1mar17 update 3mar 10am Figure 15.2 Shows that increasing the diversity of the basis set lowers The HF-SCF energy considerably, but comes nowhere near the exact experimental energy, regardless
Chemistry 14CL. Worksheet for the Molecular Modeling Workshop. (Revised FULL Version 2012 J.W. Pang) (Modified A. A. Russell)
 (Modified A. A. Russell)") Chemistry 14CL Worksheet for the Molecular Modeling Workshop (Revised FULL Version 2012 J.W. Pang) (Modified A. A. Russell) Structure of the Molecular Modeling Assignment The molecular modeling assignment
Chemistry 14CL Worksheet for the Molecular Modeling Workshop (Revised FULL Version 2012 J.W. Pang) (Modified A. A. Russell) Structure of the Molecular Modeling Assignment The molecular modeling assignment
Basis Set for Molecular Orbital Theory
 Basis Set for Molecular Orbital Theory! Different Types of Basis Functions! Different Types of Atom Center Basis Functions! Classifications of Gaussian Basis Sets! Pseudopotentials! Molecular Properties
Basis Set for Molecular Orbital Theory! Different Types of Basis Functions! Different Types of Atom Center Basis Functions! Classifications of Gaussian Basis Sets! Pseudopotentials! Molecular Properties
MO Calculation for a Diatomic Molecule. /4 0 ) i=1 j>i (1/r ij )
 i=1 j>i (1/r ij )") MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
Calculating NMR Chemical Shifts for beta-ionone O
 Calculating NMR Chemical Shifts for beta-ionone O Molecular orbital calculations can be used to get good estimates for chemical shifts. In this exercise we will calculate the chemical shifts for beta-ionone.
Calculating NMR Chemical Shifts for beta-ionone O Molecular orbital calculations can be used to get good estimates for chemical shifts. In this exercise we will calculate the chemical shifts for beta-ionone.
CHEM 344 Molecular Modeling
 CHEM 344 Molecular Modeling The Use of Computational Chemistry to Support Experimental Organic Chemistry Part 1: Molecular Orbital Theory, Hybridization, & Formal Charge * all calculation data obtained
CHEM 344 Molecular Modeling The Use of Computational Chemistry to Support Experimental Organic Chemistry Part 1: Molecular Orbital Theory, Hybridization, & Formal Charge * all calculation data obtained
Patrick: An Introduction to Medicinal Chemistry 5e MOLECULAR MODELLING EXERCISES CHAPTER 17
 MOLECULAR MODELLING EXERCISES CHAPTER 17 Exercise 17.6 Conformational analysis of n-butane Introduction Figure 1 Butane Me Me In this exercise, we will consider the possible stable conformations of butane
MOLECULAR MODELLING EXERCISES CHAPTER 17 Exercise 17.6 Conformational analysis of n-butane Introduction Figure 1 Butane Me Me In this exercise, we will consider the possible stable conformations of butane
Excited States Calculations for Protonated PAHs
 52 Chapter 3 Excited States Calculations for Protonated PAHs 3.1 Introduction Protonated PAHs are closed shell ions. Their electronic structure should therefore be similar to that of neutral PAHs, but
52 Chapter 3 Excited States Calculations for Protonated PAHs 3.1 Introduction Protonated PAHs are closed shell ions. Their electronic structure should therefore be similar to that of neutral PAHs, but
Modeling the UV-Vis Absorption of a Series of Dyes CH342L: Spectroscopy February 15, 2016
 Modeling the UV-Vis Absorption of a Series of Dyes CH342L: Spectroscopy February 15, 2016 We ll correlate the absorbance maximum of a series of dyes with structural changes between them 1. Chemicals absorb
Modeling the UV-Vis Absorption of a Series of Dyes CH342L: Spectroscopy February 15, 2016 We ll correlate the absorbance maximum of a series of dyes with structural changes between them 1. Chemicals absorb
Department of Chemistry. The Intersection of Computational Chemistry and Experiment
 Department of Chemistry The Intersection of Computational Chemistry and Experiment Structure, Vibrational and Electronic Spectra of Organic Molecules Angelo R. Rossi Department of Chemistry The University
Department of Chemistry The Intersection of Computational Chemistry and Experiment Structure, Vibrational and Electronic Spectra of Organic Molecules Angelo R. Rossi Department of Chemistry The University
Gustavus Adolphus College. Lab #5: Computational Chemistry
 CHE 372 Gustavus Adolphus College Lab #5: Computational Chemistry Introduction In this investigation we will apply the techniques of computational chemistry to several of the molecular systems that we
CHE 372 Gustavus Adolphus College Lab #5: Computational Chemistry Introduction In this investigation we will apply the techniques of computational chemistry to several of the molecular systems that we
CH342 Handin Homework 2
 CH34 Handin Homework 1. What are the quantum numbers for the energy levels that are involved in the lowest energy electronic transition for the molecule: C=C-C=C-C=C-C=C. Base your answer on the particlein
CH34 Handin Homework 1. What are the quantum numbers for the energy levels that are involved in the lowest energy electronic transition for the molecule: C=C-C=C-C=C-C=C. Base your answer on the particlein
Project 1 Report File: Chem4PB3_project_1_2017-solutions last changed: 02-Feb-2017
 Project 1 Report File: Chem4PB3_project_1_2017-solutions last changed: 02-Feb-2017 1. Formaldehyde comparison of results from different methods Yellow shaded boxes are closest to experimental Method #e-
Project 1 Report File: Chem4PB3_project_1_2017-solutions last changed: 02-Feb-2017 1. Formaldehyde comparison of results from different methods Yellow shaded boxes are closest to experimental Method #e-
计算物理作业二. Excercise 1: Illustration of the convergence of the dissociation energy for H 2 toward HF limit.
 计算物理作业二 Excercise 1: Illustration of the convergence of the dissociation energy for H 2 toward HF limit. In this exercise, basis indicates one of the following basis sets: STO-3G, cc-pvdz, cc-pvtz, cc-pvqz
计算物理作业二 Excercise 1: Illustration of the convergence of the dissociation energy for H 2 toward HF limit. In this exercise, basis indicates one of the following basis sets: STO-3G, cc-pvdz, cc-pvtz, cc-pvqz
Chemistry 4560/5560 Molecular Modeling Fall 2014
 Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Computational chemistry with GAMESS: a very brief overview with examples
 Computational chemistry with GAMESS: a very brief overview with examples PHY-6120 Molecular Physics (Spring 2015), UConn Phys. Dept. Feb 17 th 2015 H = ħ2 2μ i Intro: V(R) for diatomic molecules + k Z
Computational chemistry with GAMESS: a very brief overview with examples PHY-6120 Molecular Physics (Spring 2015), UConn Phys. Dept. Feb 17 th 2015 H = ħ2 2μ i Intro: V(R) for diatomic molecules + k Z
Exercises for Windows
 Exercises for Windows CAChe User Interface for Windows Select tool Application window Document window (workspace) Style bar Tool palette Select entire molecule Select Similar Group Select Atom tool Rotate
Exercises for Windows CAChe User Interface for Windows Select tool Application window Document window (workspace) Style bar Tool palette Select entire molecule Select Similar Group Select Atom tool Rotate
Appendix D Simulating Spectroscopic Bands Using Gaussian and PGopher
 429 Appendix D Simulating Spectroscopic Bands Using Gaussian and PGopher This appendix contains methods for using Gaussian 09 121 and PGopher 120 to simulate vibrational and electronic bands of molecules.
429 Appendix D Simulating Spectroscopic Bands Using Gaussian and PGopher This appendix contains methods for using Gaussian 09 121 and PGopher 120 to simulate vibrational and electronic bands of molecules.
Introduc)on to IQmol: Part I.!!! Shirin Faraji, Ilya Kaliman, and Anna Krylov
on to IQmol: Part I.!!! Shirin Faraji, Ilya Kaliman, and Anna Krylov") Introduc)on to IQmol: Part I!!! Shirin Faraji, Ilya Kaliman, and Anna Krylov! 1 Resources! Written by Dr. Andrew Gilbert Keep yourself up to date with IQmol website: http://iqmol.org! IQmol Youtube channel:
Introduc)on to IQmol: Part I!!! Shirin Faraji, Ilya Kaliman, and Anna Krylov! 1 Resources! Written by Dr. Andrew Gilbert Keep yourself up to date with IQmol website: http://iqmol.org! IQmol Youtube channel:
Instructions for Using Spartan 14
 Instructions for Using Spartan 14 Log in to the computer with your Colby ID and password. Click on the Spartan 14 icon in the dock at the bottom of your screen. I. Building Molecules Spartan has one main
Instructions for Using Spartan 14 Log in to the computer with your Colby ID and password. Click on the Spartan 14 icon in the dock at the bottom of your screen. I. Building Molecules Spartan has one main
Figure 1: Transition State, Saddle Point, Reaction Pathway
 Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces
Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces
This is a very succinct primer intended as supplementary material for an undergraduate course in physical chemistry.
 1 Computational Chemistry (Quantum Chemistry) Primer This is a very succinct primer intended as supplementary material for an undergraduate course in physical chemistry. TABLE OF CONTENTS Methods...1 Basis
1 Computational Chemistry (Quantum Chemistry) Primer This is a very succinct primer intended as supplementary material for an undergraduate course in physical chemistry. TABLE OF CONTENTS Methods...1 Basis
XYZ file format Protein Data Bank (pdb) file format Z - matrix
 file format Z - matrix") Chemistry block (exercise 1) In this exercise, students will be introduced how to preform simple quantum chemical calculations. Input files for Gaussian09. Output file structure. Geometry optimization,
Chemistry block (exercise 1) In this exercise, students will be introduced how to preform simple quantum chemical calculations. Input files for Gaussian09. Output file structure. Geometry optimization,
Jaguar DFT Optimizations and Transition State Searches
 Jaguar DFT Optimizations and Transition State Searches Density Functional Theory (DFT) is a quantum mechanical (QM) method that gives results superior to Hartree Fock (HF) in less computational time. A
Jaguar DFT Optimizations and Transition State Searches Density Functional Theory (DFT) is a quantum mechanical (QM) method that gives results superior to Hartree Fock (HF) in less computational time. A
QUANTUM CHEMISTRY WITH GAUSSIAN : A VERY BRIEF INTRODUCTION (PART 2)
") QUANTUM CHEMISTRY WITH GAUSSIAN : A VERY BRIEF INTRODUCTION (PART 2) TARAS V. POGORELOV AND MIKE HALLOCK SCHOOL OF CHEMICAL SCIENCES, UIUC This tutorial continues introduction to Gaussian [2]. Here we
QUANTUM CHEMISTRY WITH GAUSSIAN : A VERY BRIEF INTRODUCTION (PART 2) TARAS V. POGORELOV AND MIKE HALLOCK SCHOOL OF CHEMICAL SCIENCES, UIUC This tutorial continues introduction to Gaussian [2]. Here we
QUANTUM CHEMISTRY PROJECT 3: ATOMIC AND MOLECULAR STRUCTURE
 Chemistry 460 Fall 2017 Dr. Jean M. Standard November 1, 2017 QUANTUM CHEMISTRY PROJECT 3: ATOMIC AND MOLECULAR STRUCTURE OUTLINE In this project, you will carry out quantum mechanical calculations of
Chemistry 460 Fall 2017 Dr. Jean M. Standard November 1, 2017 QUANTUM CHEMISTRY PROJECT 3: ATOMIC AND MOLECULAR STRUCTURE OUTLINE In this project, you will carry out quantum mechanical calculations of
Conformational energy analysis
 Lab 3 Conformational energy analysis Objective This computational project deals with molecular conformations the spatial arrangement of atoms of molecules. Conformations are determined by energy, so the
Lab 3 Conformational energy analysis Objective This computational project deals with molecular conformations the spatial arrangement of atoms of molecules. Conformations are determined by energy, so the
Molecular Modeling and Conformational Analysis with PC Spartan
 Molecular Modeling and Conformational Analysis with PC Spartan Introduction Molecular modeling can be done in a variety of ways, from using simple hand-held models to doing sophisticated calculations on
Molecular Modeling and Conformational Analysis with PC Spartan Introduction Molecular modeling can be done in a variety of ways, from using simple hand-held models to doing sophisticated calculations on
Choice of Theoretical Method
 Choice of Theoretical Method 1 General Considerations PResources < Software < Computer P Expense (Time and Money) < Optimization/Single Point Calculations < Basis Set Choice < Method Choice P < Comparison
Choice of Theoretical Method 1 General Considerations PResources < Software < Computer P Expense (Time and Money) < Optimization/Single Point Calculations < Basis Set Choice < Method Choice P < Comparison
Diphenylpolyene Dye Spectra
 Diphenylpolyene Dye Spectra The purpose of this lab is to interpret uv-visible spectra of three diphenyl polyenes. The uv-visible transitions are compared to a particle-in-a-box model and to transitions
Diphenylpolyene Dye Spectra The purpose of this lab is to interpret uv-visible spectra of three diphenyl polyenes. The uv-visible transitions are compared to a particle-in-a-box model and to transitions
Ab initio calculations for potential energy surfaces. D. Talbi GRAAL- Montpellier
 Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C
 Chemistry 460 Fall 2017 Dr. Jean M. Standard November 6, 2017 QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C PART B: POTENTIAL CURVE, SPECTROSCOPIC CONSTANTS, AND DISSOCIATION ENERGY OF DIATOMIC HYDROGEN (20
Chemistry 460 Fall 2017 Dr. Jean M. Standard November 6, 2017 QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C PART B: POTENTIAL CURVE, SPECTROSCOPIC CONSTANTS, AND DISSOCIATION ENERGY OF DIATOMIC HYDROGEN (20
Transition states and reaction paths
 Transition states and reaction paths Lab 4 Theoretical background Transition state A transition structure is the molecular configuration that separates reactants and products. In a system with a single
Transition states and reaction paths Lab 4 Theoretical background Transition state A transition structure is the molecular configuration that separates reactants and products. In a system with a single
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Literature values: ΔH f, gas = % error Source: ΔH f, solid = % error. For comparison, your experimental value was ΔH f = phase:
 1 Molecular Calculations Lab: Some guideline given at the bottom of page 3. 1. Use the semi-empirical AM1 method to calculate ΔH f for the compound you used in the heat of combustion experiment. Be sure
1 Molecular Calculations Lab: Some guideline given at the bottom of page 3. 1. Use the semi-empirical AM1 method to calculate ΔH f for the compound you used in the heat of combustion experiment. Be sure
Patrick Lestrange and James B. Foresman, York College of PA C 9 C 10 C 11 C 12 O 13 H 14
 Physical Chemistry I Laboratory: Conformational Analysis of N-Boc-3-pyrrolidinol Patrick Lestrange and James B. Foresman, York College of PA C 2 C 10 C 9 O 5 C 1 C 4 C 11 C 12 N 8 C 6 C 3 O 13 O 7 H 14
Physical Chemistry I Laboratory: Conformational Analysis of N-Boc-3-pyrrolidinol Patrick Lestrange and James B. Foresman, York College of PA C 2 C 10 C 9 O 5 C 1 C 4 C 11 C 12 N 8 C 6 C 3 O 13 O 7 H 14
Chem 1140; Molecular Modeling
 P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
Introduction to Hartree-Fock calculations using ORCA and Chemcraft
 Introduction to Hartree-Fock calculations using ORCA and Chemcraft In this exercise, you will get to use software for carrying out calculations of wavefunctions for molecules, the ORCA program. While ORCA
Introduction to Hartree-Fock calculations using ORCA and Chemcraft In this exercise, you will get to use software for carrying out calculations of wavefunctions for molecules, the ORCA program. While ORCA
Tutorial I: IQ MOL and Basic DFT and MP2 Calculations 1 / 30
 Tutorial I: IQ MOL and Basic DFT and MP2 Calculations Q-Chem User Workshop, Denver March 21, 2015 1 / 30 2 / 30 Introduction to IQMOL DFT and MP2 Calculations 3 / 30 IQMOL and Q-CHEM IQMOL is an open-source
Tutorial I: IQ MOL and Basic DFT and MP2 Calculations Q-Chem User Workshop, Denver March 21, 2015 1 / 30 2 / 30 Introduction to IQMOL DFT and MP2 Calculations 3 / 30 IQMOL and Q-CHEM IQMOL is an open-source
Introduction to Ab Initio Quantum Chemical Computation
 c:\374-17\computation\computation17.doc for 9mar17 Prof. Patrik Callis 8mar17 Introduction to Ab Initio Quantum Chemical Computation Purpose: 1. To become acquainted with basic concepts of ab initio quantum
c:\374-17\computation\computation17.doc for 9mar17 Prof. Patrik Callis 8mar17 Introduction to Ab Initio Quantum Chemical Computation Purpose: 1. To become acquainted with basic concepts of ab initio quantum
The Hückel Approximation Consider a conjugated molecule i.e. a molecule with alternating double and single bonds, as shown in Figure 1.
 The Hückel Approximation In this exercise you will use a program called Hückel to look at the p molecular orbitals in conjugated molecules. The program calculates the energies and shapes of p (pi) molecular
The Hückel Approximation In this exercise you will use a program called Hückel to look at the p molecular orbitals in conjugated molecules. The program calculates the energies and shapes of p (pi) molecular
Project 3: Molecular Orbital Calculations of Diatomic Molecules. This project is worth 30 points and is due on Wednesday, May 2, 2018.
 Chemistry 362 Spring 2018 Dr. Jean M. Standard April 20, 2018 Project 3: Molecular Orbital Calculations of Diatomic Molecules In this project, you will investigate the molecular orbitals and molecular
Chemistry 362 Spring 2018 Dr. Jean M. Standard April 20, 2018 Project 3: Molecular Orbital Calculations of Diatomic Molecules In this project, you will investigate the molecular orbitals and molecular
1 An Experimental and Computational Investigation of the Dehydration of 2-Butanol
 1 An Experimental and Computational Investigation of the Dehydration of 2-Butanol Summary. 2-Butanol will be dehydrated to a mixture of 1-butene and cis- and trans-2-butene using the method described in
1 An Experimental and Computational Investigation of the Dehydration of 2-Butanol Summary. 2-Butanol will be dehydrated to a mixture of 1-butene and cis- and trans-2-butene using the method described in
Learning to Use Scigress Wagner, Eugene P. (revised May 15, 2018)
") Learning to Use Scigress Wagner, Eugene P. (revised May 15, 2018) Abstract Students are introduced to basic features of Scigress by building molecules and performing calculations on them using semi-empirical
Learning to Use Scigress Wagner, Eugene P. (revised May 15, 2018) Abstract Students are introduced to basic features of Scigress by building molecules and performing calculations on them using semi-empirical
1 Introduction to Computational Chemistry (Spartan)
") 1 Introduction to Computational Chemistry (Spartan) Start Spartan by clicking Start / Programs / Spartan Then click File / New Exercise 1 Study of H-X-H Bond Angles (Suitable for general chemistry) Structure
1 Introduction to Computational Chemistry (Spartan) Start Spartan by clicking Start / Programs / Spartan Then click File / New Exercise 1 Study of H-X-H Bond Angles (Suitable for general chemistry) Structure
Molecular simulation of copper(ii)-bound organic compounds for use in metal-organic chemical vapor deposition (MOCVD) of copper films
-bound organic compounds for use in metal-organic chemical vapor deposition (MOCVD) of copper films") Molecular simulation of copper(ii)-bound organic compounds for use in metal-organic chemical vapor deposition (MOCVD) of copper films Alexis A. Rivera-Montalvo Chemical Engineering Department University
Molecular simulation of copper(ii)-bound organic compounds for use in metal-organic chemical vapor deposition (MOCVD) of copper films Alexis A. Rivera-Montalvo Chemical Engineering Department University
Project 2. Chemistry of Transient Species in Planetary Atmospheres: Exploring the Potential Energy Surfaces of CH 2 S
 Chemistry 362 Spring 2018 Dr. Jean M. Standard March 21, 2018 Project 2. Chemistry of Transient Species in Planetary Atmospheres: Exploring the Potential Energy Surfaces of CH 2 S In this project, you
Chemistry 362 Spring 2018 Dr. Jean M. Standard March 21, 2018 Project 2. Chemistry of Transient Species in Planetary Atmospheres: Exploring the Potential Energy Surfaces of CH 2 S In this project, you
5 Electron Densities, Electrostatic Potentials, and Reactivity Indices (Spartan)
") 5 Electron Densities, Electrostatic Potentials, and Reactivity Indices (Spartan) Exercise 1 Visualizing Different Bond Types Build the H 2 molecule. Save as dihydrogen.spartan in an appropriate folder.
5 Electron Densities, Electrostatic Potentials, and Reactivity Indices (Spartan) Exercise 1 Visualizing Different Bond Types Build the H 2 molecule. Save as dihydrogen.spartan in an appropriate folder.
Practical 1: Structure and electronic properties of organic molecules. B/ Structure, electronic and vibrational properties of the water molecule
 D1CH9116 - MMCO Molecular Modelling applied to organic chemistry Practical 1: tructure and electronic properties of organic molecules B/ tructure, electronic and vibrational properties of the water molecule
D1CH9116 - MMCO Molecular Modelling applied to organic chemistry Practical 1: tructure and electronic properties of organic molecules B/ tructure, electronic and vibrational properties of the water molecule
Calculating Vibrational Spectra from Molecular Dynamics
 Calculating Vibrational Spectra from Molecular Dynamics A Simulating a Trajectory with Wannier Centers To calculate IR spectra from Molecular Dynamics, it is necessary to have dipole information for the
Calculating Vibrational Spectra from Molecular Dynamics A Simulating a Trajectory with Wannier Centers To calculate IR spectra from Molecular Dynamics, it is necessary to have dipole information for the
Computational Chemistry. An Introduction to Molecular Dynamic Simulations
 Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Technical Note Calculations of Orbital Overlap Range Function EDR( r ; d) and Overlap Distance D(r )using Multiwfn
 and Overlap Distance D(r )using Multiwfn") Technical Note Calculations of Orbital Overlap Range Function EDR( r ; d) and Overlap Distance D(r )using Multiwfn Abstract The orbital overlap range function EDR( r; d) (J. Chem. Phys. 2014, 141, 144104)
Technical Note Calculations of Orbital Overlap Range Function EDR( r ; d) and Overlap Distance D(r )using Multiwfn Abstract The orbital overlap range function EDR( r; d) (J. Chem. Phys. 2014, 141, 144104)
7 Infrared, Thermochemistry, UV-Vis, and NMR
 7 Infrared, Thermochemistry, UV-Vis, and NMR Exercise 1 Method Dependence and Scaling for the Infrared Spectrum of Formaldehyde. Build a molecule of formaldehyde using sp 2 C and atoms. Clean up the structure
7 Infrared, Thermochemistry, UV-Vis, and NMR Exercise 1 Method Dependence and Scaling for the Infrared Spectrum of Formaldehyde. Build a molecule of formaldehyde using sp 2 C and atoms. Clean up the structure
SCIGRESS Release Notes
 SCIGRESS Release Notes SCIGRESS 2.9 (3.4) General-purpose quantum-chemical package niedoida available on Windows, Linux and MacOS. New methods implemented: o Ab initio: HF, MP2. o DFT: SVWN, BLYP, B3LYP,
SCIGRESS Release Notes SCIGRESS 2.9 (3.4) General-purpose quantum-chemical package niedoida available on Windows, Linux and MacOS. New methods implemented: o Ab initio: HF, MP2. o DFT: SVWN, BLYP, B3LYP,
Computer Laboratory DFT and Frequencies
 Computer Laboratory DFT and Frequencies 1. DFT KEYWORDS FOR DFT METHODS Names for the various pure DFT models are given by combining the names for the exchange and correlation functionals. In some cases,
Computer Laboratory DFT and Frequencies 1. DFT KEYWORDS FOR DFT METHODS Names for the various pure DFT models are given by combining the names for the exchange and correlation functionals. In some cases,
Computational Material Science Part II. Ito Chao ( ) Institute of Chemistry Academia Sinica
 Institute of Chemistry Academia Sinica") Computational Material Science Part II Ito Chao ( ) Institute of Chemistry Academia Sinica Ab Initio Implementations of Hartree-Fock Molecular Orbital Theory Fundamental assumption of HF theory: each electron
Computational Material Science Part II Ito Chao ( ) Institute of Chemistry Academia Sinica Ab Initio Implementations of Hartree-Fock Molecular Orbital Theory Fundamental assumption of HF theory: each electron
Investigation 5: Infrared Spectroscopy and Molecular Modeling
 2012-13 Chemistry 120 and Chem110/IR&Modeling/Procedure 1 Investigation 5: Infrared Spectroscopy and Molecular Modeling Question: What do molecules look like and how do they move? How can we make them
2012-13 Chemistry 120 and Chem110/IR&Modeling/Procedure 1 Investigation 5: Infrared Spectroscopy and Molecular Modeling Question: What do molecules look like and how do they move? How can we make them
Chapter: 22. Visualization: Making INPUT File and Processing of Output Results
 Chapter: 22 Visualization: Making INPUT File and Processing of Output Results Keywords: visualization, input and output structure, molecular orbital, electron density. In the previous chapters, we have
Chapter: 22 Visualization: Making INPUT File and Processing of Output Results Keywords: visualization, input and output structure, molecular orbital, electron density. In the previous chapters, we have
Graphical User Interface Simplified Graphics Requests. The most commonly requested graphics (HOMO,
 Spartan 10 ( Spartan ) has been designed to address the ever increasing role that calculations play in chemistry and related fields. It represents a continued collaboration between Wavefunction, Inc.,
Spartan 10 ( Spartan ) has been designed to address the ever increasing role that calculations play in chemistry and related fields. It represents a continued collaboration between Wavefunction, Inc.,
CHE3935. Lecture 4 Quantum Mechanical Simulation Methods Continued
 CHE3935 Lecture 4 Quantum Mechanical Simulation Methods Continued 1 OUTLINE Review Introduction to CPMD MD and ensembles The functionals of density functional theory Return to ab initio methods Binding
CHE3935 Lecture 4 Quantum Mechanical Simulation Methods Continued 1 OUTLINE Review Introduction to CPMD MD and ensembles The functionals of density functional theory Return to ab initio methods Binding
The use of solvation models and the ONIOM layered approach in Gaussian.
 The use of solvation models and the NIM layered approach in Gaussian. In this lab we will consider two techniques that are very useful to model larger systems: the use of solvation models to mimic systems
The use of solvation models and the NIM layered approach in Gaussian. In this lab we will consider two techniques that are very useful to model larger systems: the use of solvation models to mimic systems
Investigation 5: Infrared Spectroscopy and Molecular Modeling
 2014 Chemistry 120 and Chem110/IR&Modeling/Procedure 1 Investigation 5: Infrared Spectroscopy and Molecular Modeling Question: What do molecules look like and how do they move? How can we make them vibrate?
2014 Chemistry 120 and Chem110/IR&Modeling/Procedure 1 Investigation 5: Infrared Spectroscopy and Molecular Modeling Question: What do molecules look like and how do they move? How can we make them vibrate?
When you download HuLiS you are requested to fill a form that tells us where HuLiS is used. It can be skipped, but we appreciate if you do so.
 User s manual Version 3.3 February 2016-04-01 S. umbel, ism2, Aix-Marseille University France For ulis 3.3 Native english teachers (or students): if you like the program but not the manual, send me corrections
User s manual Version 3.3 February 2016-04-01 S. umbel, ism2, Aix-Marseille University France For ulis 3.3 Native english teachers (or students): if you like the program but not the manual, send me corrections
Introduction to Computational Chemistry Exercise 2
 Introduction to Computational Chemistry Exercise 2 Intermolecular interactions and vibrational motion Lecturer: Antti Lignell Name Introduction In this computer exercise, we model intermolecular interactions
Introduction to Computational Chemistry Exercise 2 Intermolecular interactions and vibrational motion Lecturer: Antti Lignell Name Introduction In this computer exercise, we model intermolecular interactions
Theoretical UV/VIS Spectroscopy
 Theoretical UV/VIS Spectroscopy Why is a Ruby Red When Chromium Oxide is Green? How Does a Ruby Laser Work? Goals of this Exercise: - Calculation of the energy of electronically excited states - Understanding
Theoretical UV/VIS Spectroscopy Why is a Ruby Red When Chromium Oxide is Green? How Does a Ruby Laser Work? Goals of this Exercise: - Calculation of the energy of electronically excited states - Understanding
Name. Chem Organic Chemistry II Laboratory Exercise Molecular Modeling Part 2
 Name Chem 322 - Organic Chemistry II Laboratory Exercise Molecular Modeling Part 2 Click on Titan in the Start menu. When it boots, click on the right corner to make the window full-screen. icon in the
Name Chem 322 - Organic Chemistry II Laboratory Exercise Molecular Modeling Part 2 Click on Titan in the Start menu. When it boots, click on the right corner to make the window full-screen. icon in the
Quantum Chemical Calculations by Parallel Computer from Commodity PC Components
 Nonlinear Analysis: Modelling and Control, 2007, Vol. 12, No. 4, 461 468 Quantum Chemical Calculations by Parallel Computer from Commodity PC Components S. Bekešienė 1, S. Sėrikovienė 2 1 Institute of
Nonlinear Analysis: Modelling and Control, 2007, Vol. 12, No. 4, 461 468 Quantum Chemical Calculations by Parallel Computer from Commodity PC Components S. Bekešienė 1, S. Sėrikovienė 2 1 Institute of
AN INTRODUCTION TO QUANTUM CHEMISTRY. Mark S. Gordon Iowa State University
 AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
The Hückel Approximation
 The ückel Approximation 1 In this exercise you will use a program called ückel to look at the π molecular orbitals in conjugated molecules. The program calculates the energies and shapes of π (pi) molecular
The ückel Approximation 1 In this exercise you will use a program called ückel to look at the π molecular orbitals in conjugated molecules. The program calculates the energies and shapes of π (pi) molecular
Performance of Hartree-Fock and Correlated Methods
 Chemistry 460 Fall 2017 Dr. Jean M. Standard December 4, 2017 Performance of Hartree-Fock and Correlated Methods Hartree-Fock Methods Hartree-Fock methods generally yield optimized geomtries and molecular
Chemistry 460 Fall 2017 Dr. Jean M. Standard December 4, 2017 Performance of Hartree-Fock and Correlated Methods Hartree-Fock Methods Hartree-Fock methods generally yield optimized geomtries and molecular
Basis Sets and Basis Set Notation
 Chemistry 46 Fall 215 Dr. Jean M. Standard November 29, 217 Basis Sets and Basis Set Notation Using the LCAO-MO approximation, molecular orbitals can be represented as linear combinations of atomic orbitals,
Chemistry 46 Fall 215 Dr. Jean M. Standard November 29, 217 Basis Sets and Basis Set Notation Using the LCAO-MO approximation, molecular orbitals can be represented as linear combinations of atomic orbitals,
Lectures Spectroscopy. Fall 2012
 Lectures 19-20 Spectroscopy Fall 2012 1 spectroscopic principles (Chem 1M/1N exps. #6 and #11) 4 spectroscopic excitations ( E = h = hc/ ; = c ) (nm) (sec -1 ) radiation technique molecular excitation
Lectures 19-20 Spectroscopy Fall 2012 1 spectroscopic principles (Chem 1M/1N exps. #6 and #11) 4 spectroscopic excitations ( E = h = hc/ ; = c ) (nm) (sec -1 ) radiation technique molecular excitation
Hints on Using the Orca Program
 Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Hints on Using the Orca Program
Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Hints on Using the Orca Program
( R)Ψ el ( r;r) = E el ( R)Ψ el ( r;r)
Ψ el ( r;r) = E el ( R)Ψ el ( r;r)") Born Oppenheimer Approximation: Ĥ el ( R)Ψ el ( r;r) = E el ( R)Ψ el ( r;r) For a molecule with N electrons and M nuclei: Ĥ el What is E el (R)? s* potential surface Reaction Barrier Unstable intermediate
Born Oppenheimer Approximation: Ĥ el ( R)Ψ el ( r;r) = E el ( R)Ψ el ( r;r) For a molecule with N electrons and M nuclei: Ĥ el What is E el (R)? s* potential surface Reaction Barrier Unstable intermediate
Semi-Empirical MO Methods
 Semi-Empirical MO Methods the high cost of ab initio MO calculations is largely due to the many integrals that need to be calculated (esp. two electron integrals) semi-empirical MO methods start with the
Semi-Empirical MO Methods the high cost of ab initio MO calculations is largely due to the many integrals that need to be calculated (esp. two electron integrals) semi-empirical MO methods start with the
Molecular Simulation I
 Molecular Simulation I Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular Simulation I Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Raman Spectroscopy of Liquids
 Chemistry 357 Spring 2013 Raman Spectroscopy of Liquids Lab TA: Paul Dent pwdent@syr.edu PURPOSE: You will investigate Raman light scattering of several different molecular liquids. You will also determine
Chemistry 357 Spring 2013 Raman Spectroscopy of Liquids Lab TA: Paul Dent pwdent@syr.edu PURPOSE: You will investigate Raman light scattering of several different molecular liquids. You will also determine
Athena Visual Software, Inc. 1
 Athena Visual Studio Visual Kinetics Tutorial VisualKinetics is an integrated tool within the Athena Visual Studio software environment, which allows scientists and engineers to simulate the dynamic behavior
Athena Visual Studio Visual Kinetics Tutorial VisualKinetics is an integrated tool within the Athena Visual Studio software environment, which allows scientists and engineers to simulate the dynamic behavior
Same idea for polyatomics, keep track of identical atom e.g. NH 3 consider only valence electrons F(2s,2p) H(1s)
 H(1s)") XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
NMR Predictor. Introduction
 NMR Predictor This manual gives a walk-through on how to use the NMR Predictor: Introduction NMR Predictor QuickHelp NMR Predictor Overview Chemical features GUI features Usage Menu system File menu Edit
NMR Predictor This manual gives a walk-through on how to use the NMR Predictor: Introduction NMR Predictor QuickHelp NMR Predictor Overview Chemical features GUI features Usage Menu system File menu Edit
Session 7 Overview: Part A I. Prediction of Vibrational Frequencies (IR) Part B III. Prediction of Electronic Transitions (UV-Vis) IV.
 Part B III. Prediction of Electronic Transitions (UV-Vis) IV.") Session 7 Overview: Part A I. Prediction of Vibrational Frequencies (IR) II. Thermochemistry Part B III. Prediction of Electronic Transitions (UV-Vis) IV. NMR Predictions 1 I. Prediction of Vibrational
Session 7 Overview: Part A I. Prediction of Vibrational Frequencies (IR) II. Thermochemistry Part B III. Prediction of Electronic Transitions (UV-Vis) IV. NMR Predictions 1 I. Prediction of Vibrational