Project 1 Report File: Chem4PB3_project_1_2017-solutions last changed: 02-Feb-2017
|
|
|
- Karen Simon
- 6 years ago
- Views:
Transcription
1 Project 1 Report File: Chem4PB3_project_1_2017-solutions last changed: 02-Feb Formaldehyde comparison of results from different methods Yellow shaded boxes are closest to experimental Method #e- DFT Experimental Property AM1 MM - UFF HF STO- 3G HF STO-3G forced C 2v RHF 6311G forced C 2v B3LYP forced C 2v E (ev), r (pm) wavenumbers method AM1 UFF RHF RHF RHF DFT - B3LYP - Basis set minimal none STO-3G STO-3G 6-311G 6-311G - # basis functions # iterations CPU time (s) Energy (hartree) (*) Dipole Moment (debye) Reported symmetry Cs Cs Cs C2v C2v C2v C2v R(C=O) (pm) R(C-H) (pm) φ(h-c=o) ( o ) φ(h-c-h) ( o ) Torsion (planarity) ( o ) O 1s a C 1s a O 2s a C 2s a σ(c-h) b σ(c-h) a π(c=o) b O LP/CH b π*(c=o) - b σ*(c-h) σ*(c-h) σ*(c=o) CH2 s-str a (IR, R) CO str a (IR) CH2 scis a (IR, R) CH2 a-str b (IR) CH2 rock b (IR) CH2 wag b (IR
2 Formaldehyde ground state DETAILS of the calculations AM1 ZDO basis set (*) computes valence electrons only Geometry : Cs E hartree (1 hartree = ev) Imaginary freq: 0 Dipole moment: Molecular Mechanics used UFF force field Amber did not produce results No electronic structure information, only geometry & Thermochemistry HF- small basis set: restricted, STO-3G basis Found I only ran it with frequency, not optimization Re-ran using CLEAN, and POINT GROUP = C2v. Did not run formaldehyde - HFR STO-3G Redundant internal coordinates taken from checkpoint file: formaldehyde-hfr-sto-3g.chk Charge = 0 Multiplicity = 1 C,0, , ,0. O,0, , ,0. H,0, , , H,0, , , WANTED AN INTEGER AS INPUT. FOUND A STRING AS INPUT. formaldehyde - HFR STO-3G STRANGE - the com file looked sensible - could not see a non-numerical value Problem was the additional keywords I had included, genchk in particular (that allows restart but only works AFTER you have one valid calculation to generate the first *.chk) 2
3 HF large basis set (6-311G) There are now way more (8 occ, 24 unocc) basis functions than the minimal basis description used to generate the simple LCAO-MO picture. The (extra) unoccupied orbitals are usually viewed as meaningless by the ab initio QM community. For spectroscopists however, they can be useful to describe the Rydberg excited states. In general they are fluffier orbitals than those that come out of the STO-3G minimal basis description. You need to look at the MO pictures to map specific ones of the 24 unocc. MOs in the 6311G picture to the 4 unocc. MOs of the minimal basis description. Orbital symmetries: Occupied (A1) (A1) (A1) (A1) (B2) (A1) (B1) (B2) Virtual (B1) (A1) (B2) (A1) (A1) (B1) (B2) (B2) (A1) (A1) (A1) (B1) (B2) (A1) (A1) (B2) (B1) (A1) (B2) (B1) (B2) (A1) (A1) (A1) The electronic state is 1-A1. Alpha occ. eigenvalues Alpha occ. eigenvalues Alpha virt. eigenvalues Alpha virt. eigenvalues Alpha virt. eigenvalues Alpha virt. eigenvalues Alpha virt. eigenvalues Some general comments 1. AM1 is by FAR the best at computing orbital energies that agree with PES IPs, within Koopman s Theorem. Perhaps that is no surprise as AM1 is most likely parametrized based on matching to valence IPs as well as other data. See for details on how the parameters in various semi-empirical codes were established. 3
4 AM AM1 Predicted vs. actual Vib. Freq. Calculated (ev) STO-3G HF 6311G DFT Calculated STO-3G HF-6311G DFT 1500 Calculated vs. experimental IPs Experimental (ev) Experimental Experimental data source of GEOMETRY, dipole moment: Sources of photoelectron spectra & IPs (a) XPS K. Kuramoto, M. Ehara, H. Nakatsujia, M. Kitajima, H. Tanaka, A. De Fanis, Y. Tamenori, K. Ueda J. El. Spec 142 (2005) (b) PES He(II) M.P. Kean, S. Lunell, A. Naves de Brito, M. Carlsson-Gothea, S. Svensson, B. Wannberg and L. Karlsson, J. El. Spec 56 (1991) (c) PES He(I) - A. D. Baker, C. Barer, C.R. Brundle and D. W. Turner,, Int. J. Mass Spec 1 (1968) 285 Adiabatic 10.88(4) ev, 14.09(5) ev, 15.85(4) ev, and 16.25(4) ev, ~20.5 Vertical 10.88(4) ev, 14.38(8)eV, 16.00(9)eV, and ev 4




")
 (&")



")
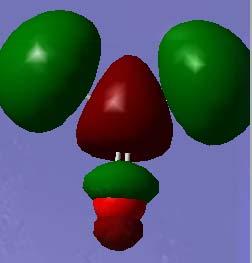
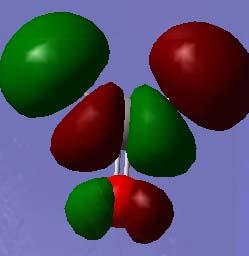

5 Orbital plots (HF STO-3G) O 1s a1 C 1s a1 O 2s a1 C 2s a1 Pi & C-H sig (C-H) (σ*(c=o) π(c=o) σ(c-h) (& O lone pair using the default e- threshold π*(c=o) σ*(c-h) σ*(c-h) σ*(c=o) 5
 For the HF (STO-3G), HF (6-311G) and DFT calculations, using the TDSCF energy method gives Excited state AM-1 HF-STO-3G HF-6-311G DFT BL3LYP EXPERIMENT 1 symmetry A2 A2 A2 A2 Energy (ev) 2.")
6 1b. Electronic (UV-vis) Spectroscopy (Ann. Rev. Phys. Chem : 31-58) For the HF (STO-3G), HF (6-311G) and DFT calculations, using the TDSCF energy method gives Excited state AM-1 HF-STO-3G HF-6-311G DFT BL3LYP EXPERIMENT 1 symmetry A2 A2 A2 A2 Energy (ev) Intensity (osc str) Dominant config 6 7 (0.705) NB add 2 to match orb# 5 -> 9 (.12) 8 -> 9 (.70) 8 -> 9 (0.70) 8 -> 9 (0.70) 2 symmetry B1 B1 B1 B1 Energy (ev) Intensity (osc str) Dominant config 6 -> 8 (.61) 6 -> 9 (.71) 6 -> 9 (.70) 6 -> 9 (.70) 3 symmetry A1 A1 B2 B2 Energy (ev) Intensity (osc str) Dominant config 6 -> 9 (0.62) 6 -> 12 (.18) 8 -> 10 (0.69) 8 -> 10 (0.71) 7 -> 9 (.68) AM-1 HF STO-3G E in ev HF 6-311G DFT BL3LyP 6-311G STO-3G output Excited State 1: Singlet-A ev nm f= <S**2>= > > Homo (O LP) Lumo (π* C=O ) A 1 A 2 forbidden in C 2v Excited State 2: Singlet-B ev nm f= <S**2>= > σ(c-h) a1 Lumo (π*) Excited State 3: Singlet-A ev nm f= <S**2>= > > π C=O a1 Lumo (π*) Formaldehyde is a common preservative in cosmetics, but this compound is also known as an irritant agent that can cause contact allergy [1]. Formaldehyde is frequently used in water-based formulations such as shampoo, conditioner, shower/bath gel, liquid hand wash. [2]. Even products designed for children such as bubble bath or baby shampoo include formaldehyde in their composition. (Analytica Chimica Acta 674 (2010) 59 63) 6
7 7
 Total energy (kj/mol) Total E (10 5 kj/mol) 1,3-Butadiene - cis 0.0505507 132.7-1,3-Butadiene - trans 0.0493885 129.7-4.005 Ethene 0.0264660 69.")
8 2. Diels-Alder reaction PM3 semiempirical calculation Ground state Energies 1 atomic unit (or Hartree) equals kj/mol pm3 HF 3-21G Molecule Total energy (Hartree) Total energy (kj/mol) Total E (10 5 kj/mol) 1,3-Butadiene - cis ,3-Butadiene - trans Ethene REACTANTS Cyclohexene Transition state
 For butadiene there are multiple SCF done cycles have to get to the LAST ONE to get final optimized energy")
9 Cis-Butadiene followed the instructions in the Harvey website NB the starting geometry was not carefully done. Perhaps should do an MM optimization first? (Spartan had a clean-up procedure in its input process; Gaussian has the same Edit~Clean ) For butadiene there are multiple SCF done cycles have to get to the LAST ONE to get final optimized energy line 248/4645 SCF Done: E(RPM3) = A.U. after 20 cycles line 479/4645 SCF Done: E(RPM3) = A.U. after 16 cycles line 652/4645 SCF Done: E(RPM3) = A.U. after 16 cycles line 826/4645 SCF Done: E(RPM3) = A.U. after 14 cycles line 998/4645 SCF Done: E(RPM3) = A.U. after 13 cycles line 1172/4645 SCF Done: E(RPM3) = A.U. after 12 cycles geometry restored to original line 1343/4645 SCF Done: E(RPM3) = E-01 A.U. after 12 cycles.... line 4371/4645 SCF Done: E(RPM3) = E-01 A.U. after 13 cycles ============ used clean, and then the default input file, not the Harvey modified one line 248/1774 SCF Done: E(RPM3) = E-01 A.U. after 12 cycles... line 1503/1774 SCF Done: E(RPM3) = E-01 A.U. after 13 cycles ============ using the Harvey format, get same answer line 1503/1774 SCF Done: E(RPM3) = E-01 A.U. after 13 cycles ======================== Cis-Butadiene Trans butadiene ======================== 9

![77 kj/mol] Thus pm3 method over-estimates the Thermochemistry by ~*4 ======================== Ethene SCF Done: E(RPM3) = 0.](/docs-images/71/65846055/images/10-1.jpg "264660251344E-01 A.U. after 7 cycles ======================== Cyclohexene SCF Done: E(RPM3) = -0.")
10 Trans butadiene line 993/1278 SCF Done: E(RPM3) = E-01 A.U. after 13 cycles Energy difference (cis trans) = 3.05 kj/mol [literature says trans is more stable than cis by 3.21 kcal/mol, or 0.77 kj/mol] Thus pm3 method over-estimates the Thermochemistry by ~*4 ======================== Ethene SCF Done: E(RPM3) = E-01 A.U. after 7 cycles ======================== Cyclohexene SCF Done: E(RPM3) = E-02 ======================== Diels Alder Thermochemistry 10
 structure")


 = - 934 cm -1 Movie made of")
11 ============= using the supplied hint geometry, but had to rebond in a sensible manner 1st try, I used just OPT & TS (Berny) structure reverted to two non-bonded molecules with delocalized bonding in the butadiene 2 nd try, I added partial bonds to make the D-A adduct, and then used OPT&FREQ line 3212/4218 SCF Done: E(RPM3) = A.U. after 2 cycles is LAST of the SCF Done imaginary frequency (the ethane moving against the cis butadiene) = cm -1 Movie made of the motion along the reaction co-ordinate (the imaginary frequency) 11
12 3. nmr calculation for pyrazinamide used RHF/6-31G calculations (forgot the d ) from my RHF/6-31G calc nucleus Chem. Shift ppm wrt ref. Calc (*) isotropic anisotropy C C C C C H H H H H (*) Chis et al. Chem. Phys 316 (2005) 153 ; B3LYP/6-31G(d) for C-1 monomer (see below) My geometry 1 C C C C H H H N N N C O H H

 153).")

13 C nmr H nmr The poor agreement of the predicted 1 H chemical shifts for H13 and H14 with experiment are because this molecule likes to dimerize by forming H-bonds between H14-O9 (see V. Chis et al. Chemical Phys. 316 (2005) 153). Note that the Chis et al calculations also show BOTH H-13 and H-14 too low, even though only H-14 is implicated in the H-bonding. 13
CHEM 344 Molecular Modeling
 CHEM 344 Molecular Modeling The Use of Computational Chemistry to Support Experimental Organic Chemistry Part 1: Molecular Orbital Theory, Hybridization, & Formal Charge * all calculation data obtained
CHEM 344 Molecular Modeling The Use of Computational Chemistry to Support Experimental Organic Chemistry Part 1: Molecular Orbital Theory, Hybridization, & Formal Charge * all calculation data obtained
Computational Chemistry Using the University of Alaska WebMO Site
 2/7/2017 1 Computational Chemistry Using the University of Alaska WebMO Site John Keller Department of Chemistry & Biochemistry University of Alaska Fairbanks Intro and operation of WebMO and MOPAC Basic
2/7/2017 1 Computational Chemistry Using the University of Alaska WebMO Site John Keller Department of Chemistry & Biochemistry University of Alaska Fairbanks Intro and operation of WebMO and MOPAC Basic
one ν im: transition state saddle point
 Hypothetical Potential Energy Surface Ethane conformations Hartree-Fock theory, basis set stationary points all ν s >0: minimum eclipsed one ν im: transition state saddle point multiple ν im: hilltop 1
Hypothetical Potential Energy Surface Ethane conformations Hartree-Fock theory, basis set stationary points all ν s >0: minimum eclipsed one ν im: transition state saddle point multiple ν im: hilltop 1
Molecular Simulation I
 Molecular Simulation I Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular Simulation I Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
IFM Chemistry Computational Chemistry 2010, 7.5 hp LAB2. Computer laboratory exercise 1 (LAB2): Quantum chemical calculations
: Quantum chemical calculations") Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Advanced Electronic Structure Theory Density functional theory. Dr Fred Manby
 Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
Using Web-Based Computations in Organic Chemistry
 10/30/2017 1 Using Web-Based Computations in Organic Chemistry John Keller UAF Department of Chemistry & Biochemistry The UAF WebMO site Practical aspects of computational chemistry theory and nomenclature
10/30/2017 1 Using Web-Based Computations in Organic Chemistry John Keller UAF Department of Chemistry & Biochemistry The UAF WebMO site Practical aspects of computational chemistry theory and nomenclature
Density Functional Theory
 Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Session 7 Overview: Part A I. Prediction of Vibrational Frequencies (IR) Part B III. Prediction of Electronic Transitions (UV-Vis) IV.
 Part B III. Prediction of Electronic Transitions (UV-Vis) IV.") Session 7 Overview: Part A I. Prediction of Vibrational Frequencies (IR) II. Thermochemistry Part B III. Prediction of Electronic Transitions (UV-Vis) IV. NMR Predictions 1 I. Prediction of Vibrational
Session 7 Overview: Part A I. Prediction of Vibrational Frequencies (IR) II. Thermochemistry Part B III. Prediction of Electronic Transitions (UV-Vis) IV. NMR Predictions 1 I. Prediction of Vibrational
Conjugated Systems. With conjugated double bonds resonance structures can be drawn
 Conjugated Systems Double bonds in conjugation behave differently than isolated double bonds With conjugated double bonds resonance structures can be drawn With isolated double bonds cannot draw resonance
Conjugated Systems Double bonds in conjugation behave differently than isolated double bonds With conjugated double bonds resonance structures can be drawn With isolated double bonds cannot draw resonance
Chemistry 5021/8021 Computational Chemistry 3/4 Credits Spring Semester 2000 ( Due 4 / 3 / 00 )
") Chemistry 5021/8021 Computational Chemistry 3/4 Credits Spring Semester 2000 ( Due 4 / 3 / 00 ) This problem set will take longer than the last one in the sense that you may need to submit some jobs, leave,
Chemistry 5021/8021 Computational Chemistry 3/4 Credits Spring Semester 2000 ( Due 4 / 3 / 00 ) This problem set will take longer than the last one in the sense that you may need to submit some jobs, leave,
Ethene. Introduction. The ethene molecule is planar (i.e. all the six atoms lie in the same plane) and has a high degree of symmetry:
 and has a high degree of symmetry:") FY1006 Innføring i kvantefysikk og TFY4215 Kjemisk fysikk og kvantemekanikk Spring 2012 Chemical Physics Exercise 1 To be delivered by Friday 27.04.12 Introduction Ethene. Ethylene, C 2 H 4, or ethene,
FY1006 Innføring i kvantefysikk og TFY4215 Kjemisk fysikk og kvantemekanikk Spring 2012 Chemical Physics Exercise 1 To be delivered by Friday 27.04.12 Introduction Ethene. Ethylene, C 2 H 4, or ethene,
Chemistry 4021/8021 Computational Chemistry 3/4 Credits Spring Semester 2007 Key PS3
 Chemistry 4021/8021 Computational Chemistry 3/4 Credits Spring Semester 2007 Key PS3 1. Below are two isomeric geometries that we previously examined in Problem Sets 1 and 2 as both C 10 H 16 and Si 10
Chemistry 4021/8021 Computational Chemistry 3/4 Credits Spring Semester 2007 Key PS3 1. Below are two isomeric geometries that we previously examined in Problem Sets 1 and 2 as both C 10 H 16 and Si 10
计算物理作业二. Excercise 1: Illustration of the convergence of the dissociation energy for H 2 toward HF limit.
 计算物理作业二 Excercise 1: Illustration of the convergence of the dissociation energy for H 2 toward HF limit. In this exercise, basis indicates one of the following basis sets: STO-3G, cc-pvdz, cc-pvtz, cc-pvqz
计算物理作业二 Excercise 1: Illustration of the convergence of the dissociation energy for H 2 toward HF limit. In this exercise, basis indicates one of the following basis sets: STO-3G, cc-pvdz, cc-pvtz, cc-pvqz
Practical 1: Structure and electronic properties of organic molecules. B/ Structure, electronic and vibrational properties of the water molecule
 D1CH9116 - MMCO Molecular Modelling applied to organic chemistry Practical 1: tructure and electronic properties of organic molecules B/ tructure, electronic and vibrational properties of the water molecule
D1CH9116 - MMCO Molecular Modelling applied to organic chemistry Practical 1: tructure and electronic properties of organic molecules B/ tructure, electronic and vibrational properties of the water molecule
Same idea for polyatomics, keep track of identical atom e.g. NH 3 consider only valence electrons F(2s,2p) H(1s)
 H(1s)") XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
Introduction to Hartree-Fock calculations in Spartan
 EE5 in 2008 Hannes Jónsson Introduction to Hartree-Fock calculations in Spartan In this exercise, you will get to use state of the art software for carrying out calculations of wavefunctions for molecues,
EE5 in 2008 Hannes Jónsson Introduction to Hartree-Fock calculations in Spartan In this exercise, you will get to use state of the art software for carrying out calculations of wavefunctions for molecues,
Figure 1: Transition State, Saddle Point, Reaction Pathway
 Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces
Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces
Hints on Using the Orca Program
 Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Hints on Using the Orca Program
Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Hints on Using the Orca Program
Department of Chemistry. The Intersection of Computational Chemistry and Experiment
 Department of Chemistry The Intersection of Computational Chemistry and Experiment Structure, Vibrational and Electronic Spectra of Organic Molecules Angelo R. Rossi Department of Chemistry The University
Department of Chemistry The Intersection of Computational Chemistry and Experiment Structure, Vibrational and Electronic Spectra of Organic Molecules Angelo R. Rossi Department of Chemistry The University
Jack Smith. Center for Environmental, Geotechnical and Applied Science. Marshall University
 Jack Smith Center for Environmental, Geotechnical and Applied Science Marshall University -- Division of Science and Research WV Higher Education Policy Commission WVU HPC Summer Institute June 20, 2014
Jack Smith Center for Environmental, Geotechnical and Applied Science Marshall University -- Division of Science and Research WV Higher Education Policy Commission WVU HPC Summer Institute June 20, 2014
Quantum Chemical Simulations and Descriptors. Dr. Antonio Chana, Dr. Mosè Casalegno
 Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Literature values: ΔH f, gas = % error Source: ΔH f, solid = % error. For comparison, your experimental value was ΔH f = phase:
 1 Molecular Calculations Lab: Some guideline given at the bottom of page 3. 1. Use the semi-empirical AM1 method to calculate ΔH f for the compound you used in the heat of combustion experiment. Be sure
1 Molecular Calculations Lab: Some guideline given at the bottom of page 3. 1. Use the semi-empirical AM1 method to calculate ΔH f for the compound you used in the heat of combustion experiment. Be sure
NMR and IR spectra & vibrational analysis
 Lab 5: NMR and IR spectra & vibrational analysis A brief theoretical background 1 Some of the available chemical quantum methods for calculating NMR chemical shifts are based on the Hartree-Fock self-consistent
Lab 5: NMR and IR spectra & vibrational analysis A brief theoretical background 1 Some of the available chemical quantum methods for calculating NMR chemical shifts are based on the Hartree-Fock self-consistent
Chemistry 416 Spectroscopy Fall Semester 1997 Dr. Rainer Glaser
 Chemistry 416 Spectroscopy Fall Semester 1997 Dr. Rainer Glaser Third 1-Hour Examination Vibrational Spectroscopy Monday, November 24, 1997, 8:40-9:30 Name: Answer Key Question 1 (Combination) 25 Question
Chemistry 416 Spectroscopy Fall Semester 1997 Dr. Rainer Glaser Third 1-Hour Examination Vibrational Spectroscopy Monday, November 24, 1997, 8:40-9:30 Name: Answer Key Question 1 (Combination) 25 Question
Chem 1140; Molecular Modeling
 P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
John Keller Department of Chemistry & Biochemistry University of Alaska Fairbanks
 10/15/2016 1 WebMO & Gaussian John Keller Department of Chemistry & Biochemistry University of Alaska Fairbanks Corrections and updates 9-5-2017 SCHEDULE 9-10 Intro and basic operation of WebMO and MOPAC
10/15/2016 1 WebMO & Gaussian John Keller Department of Chemistry & Biochemistry University of Alaska Fairbanks Corrections and updates 9-5-2017 SCHEDULE 9-10 Intro and basic operation of WebMO and MOPAC
Chemistry 433 Computational Chemistry Fall Semester 2002 Dr. Rainer Glaser
 Chemistry 433 Computational Chemistry Fall Semester 2002 Dr. Rainer Glaser Second 1-Hour Examination Electron Correlation Methods Wednesday, November 6, 2002, 11:00-11:50 Name: Answer Key Question 1. Electron
Chemistry 433 Computational Chemistry Fall Semester 2002 Dr. Rainer Glaser Second 1-Hour Examination Electron Correlation Methods Wednesday, November 6, 2002, 11:00-11:50 Name: Answer Key Question 1. Electron
Molecular Orbital Theory. Molecular Orbital Theory: Electrons are located in the molecule, not held in discrete regions between two bonded atoms
 Molecular Orbital Theory Valence Bond Theory: Electrons are located in discrete pairs between specific atoms Molecular Orbital Theory: Electrons are located in the molecule, not held in discrete regions
Molecular Orbital Theory Valence Bond Theory: Electrons are located in discrete pairs between specific atoms Molecular Orbital Theory: Electrons are located in the molecule, not held in discrete regions
Conformational energy analysis
 Lab 3 Conformational energy analysis Objective This computational project deals with molecular conformations the spatial arrangement of atoms of molecules. Conformations are determined by energy, so the
Lab 3 Conformational energy analysis Objective This computational project deals with molecular conformations the spatial arrangement of atoms of molecules. Conformations are determined by energy, so the
Chemistry 4560/5560 Molecular Modeling Fall 2014
 Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
CHAPTER 9 THEORY OF RESONANCE BY, G.DEEPA
 CHAPTER 9 THEORY OF RESONANCE BY, G.DEEPA Conjugation in Alkadienes and Allylic Systems conjugation a series of overlapping p orbitals The Allyl Group allylic position is the next to a double bond 1 allyl
CHAPTER 9 THEORY OF RESONANCE BY, G.DEEPA Conjugation in Alkadienes and Allylic Systems conjugation a series of overlapping p orbitals The Allyl Group allylic position is the next to a double bond 1 allyl
Be H. Delocalized Bonding. Localized Bonding. σ 2. σ 1. Two (sp-1s) Be-H σ bonds. The two σ bonding MO s in BeH 2. MO diagram for BeH 2
 Be-H σ bonds. The two σ bonding MO s in BeH 2. MO diagram for BeH 2") The Delocalized Approach to Bonding: The localized models for bonding we have examined (Lewis and VBT) assume that all electrons are restricted to specific bonds between atoms or in lone pairs. In contrast,
The Delocalized Approach to Bonding: The localized models for bonding we have examined (Lewis and VBT) assume that all electrons are restricted to specific bonds between atoms or in lone pairs. In contrast,
CHEM 2010 Symmetry, Electronic Structure and Bonding Winter Numbering of Chapters and Assigned Problems
 CHEM 2010 Symmetry, Electronic Structure and Bonding Winter 2011 Numbering of Chapters and Assigned Problems The following table shows the correspondence between the chapter numbers in the full book (Physical
CHEM 2010 Symmetry, Electronic Structure and Bonding Winter 2011 Numbering of Chapters and Assigned Problems The following table shows the correspondence between the chapter numbers in the full book (Physical
Molecular Orbitals for Ozone
 Molecular Orbitals for Ozone Purpose: In this exercise you will do semi-empirical molecular orbital calculations on ozone with the goal of understanding the molecular orbital print out provided by Spartan
Molecular Orbitals for Ozone Purpose: In this exercise you will do semi-empirical molecular orbital calculations on ozone with the goal of understanding the molecular orbital print out provided by Spartan
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
MO Calculation for a Diatomic Molecule. /4 0 ) i=1 j>i (1/r ij )
 i=1 j>i (1/r ij )") MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
Chemistry 433 Computational Chemistry Fall Semester 2002 Dr. Rainer Glaser
 Chemistry 433 Computational Chemistry Fall Semester 2002 Dr. Rainer Glaser Second 1-Hour Examination Electron Correlation Methods Wednesday, November 6, 2002, 11:00-11:50 Name: Question 1. Electron Correlation.
Chemistry 433 Computational Chemistry Fall Semester 2002 Dr. Rainer Glaser Second 1-Hour Examination Electron Correlation Methods Wednesday, November 6, 2002, 11:00-11:50 Name: Question 1. Electron Correlation.
Semi-Empirical MO Methods
 Semi-Empirical MO Methods the high cost of ab initio MO calculations is largely due to the many integrals that need to be calculated (esp. two electron integrals) semi-empirical MO methods start with the
Semi-Empirical MO Methods the high cost of ab initio MO calculations is largely due to the many integrals that need to be calculated (esp. two electron integrals) semi-empirical MO methods start with the
Chapter 14: Conjugated Dienes
 Chapter 14: Conjugated Dienes Coverage: 1. Conjugated vs Nonconjugated dienes and Stability 2. MO picture of 1,3-butadiene 3. Electrophilic addition to Dienes 4. Kinetic vs Thermodynamic Control 5. Diels-Alder
Chapter 14: Conjugated Dienes Coverage: 1. Conjugated vs Nonconjugated dienes and Stability 2. MO picture of 1,3-butadiene 3. Electrophilic addition to Dienes 4. Kinetic vs Thermodynamic Control 5. Diels-Alder
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Transition states and reaction paths
 Transition states and reaction paths Lab 4 Theoretical background Transition state A transition structure is the molecular configuration that separates reactants and products. In a system with a single
Transition states and reaction paths Lab 4 Theoretical background Transition state A transition structure is the molecular configuration that separates reactants and products. In a system with a single
Supplementary Information
 Supplementary Information Cobalt analogues of Roussin's red salt esters: a density functional study Menyhárt B. Sárosi and R. Bruce King* Table S1. BP86/6-311G(d) electronic energies and thermodynamic
Supplementary Information Cobalt analogues of Roussin's red salt esters: a density functional study Menyhárt B. Sárosi and R. Bruce King* Table S1. BP86/6-311G(d) electronic energies and thermodynamic
1 Introduction to Computational Chemistry (Spartan)
") 1 Introduction to Computational Chemistry (Spartan) Start Spartan by clicking Start / Programs / Spartan Then click File / New Exercise 1 Study of H-X-H Bond Angles (Suitable for general chemistry) Structure
1 Introduction to Computational Chemistry (Spartan) Start Spartan by clicking Start / Programs / Spartan Then click File / New Exercise 1 Study of H-X-H Bond Angles (Suitable for general chemistry) Structure
QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C
 Chemistry 460 Fall 2017 Dr. Jean M. Standard November 6, 2017 QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C PART B: POTENTIAL CURVE, SPECTROSCOPIC CONSTANTS, AND DISSOCIATION ENERGY OF DIATOMIC HYDROGEN (20
Chemistry 460 Fall 2017 Dr. Jean M. Standard November 6, 2017 QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C PART B: POTENTIAL CURVE, SPECTROSCOPIC CONSTANTS, AND DISSOCIATION ENERGY OF DIATOMIC HYDROGEN (20
Introduction to Ab Initio Quantum Chemical Computation
 c:\374-17\computation\computation17.doc for 9mar17 Prof. Patrik Callis 8mar17 Introduction to Ab Initio Quantum Chemical Computation Purpose: 1. To become acquainted with basic concepts of ab initio quantum
c:\374-17\computation\computation17.doc for 9mar17 Prof. Patrik Callis 8mar17 Introduction to Ab Initio Quantum Chemical Computation Purpose: 1. To become acquainted with basic concepts of ab initio quantum
LUMO + 1 LUMO. Tómas Arnar Guðmundsson Report 2 Reikniefnafræði G
 Q1: Display all the MOs for N2 in your report and classify each one of them as bonding, antibonding or non-bonding, and say whether the symmetry of the orbital is σ or π. Sketch a molecular orbital diagram
Q1: Display all the MOs for N2 in your report and classify each one of them as bonding, antibonding or non-bonding, and say whether the symmetry of the orbital is σ or π. Sketch a molecular orbital diagram
The symmetry properties & relative energies of atomic orbitals determine how they react to form molecular orbitals. These molecular orbitals are then
 1 The symmetry properties & relative energies of atomic orbitals determine how they react to form molecular orbitals. These molecular orbitals are then filled with the available electrons according to
1 The symmetry properties & relative energies of atomic orbitals determine how they react to form molecular orbitals. These molecular orbitals are then filled with the available electrons according to
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
International Journal of Materials Science ISSN Volume 12, Number 2 (2017) Research India Publications
 Research India Publications") HF, DFT Computations and Spectroscopic study of Vibrational frequency, HOMO-LUMO Analysis and Thermodynamic Properties of Alpha Bromo Gamma Butyrolactone K. Rajalakshmi 1 and A.Susila 2 1 Department of
HF, DFT Computations and Spectroscopic study of Vibrational frequency, HOMO-LUMO Analysis and Thermodynamic Properties of Alpha Bromo Gamma Butyrolactone K. Rajalakshmi 1 and A.Susila 2 1 Department of
DFT calculations of NMR indirect spin spin coupling constants
 DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Lecture 4: Band theory
 Lecture 4: Band theory Very short introduction to modern computational solid state chemistry Band theory of solids Molecules vs. solids Band structures Analysis of chemical bonding in Reciprocal space
Lecture 4: Band theory Very short introduction to modern computational solid state chemistry Band theory of solids Molecules vs. solids Band structures Analysis of chemical bonding in Reciprocal space
with the larger dimerization energy also exhibits the larger structural changes.
 A7. Looking at the image and table provided below, it is apparent that the monomer and dimer are structurally almost identical. Although angular and dihedral data were not included, these data are also
A7. Looking at the image and table provided below, it is apparent that the monomer and dimer are structurally almost identical. Although angular and dihedral data were not included, these data are also
Theoretical Chemistry - Level II - Practical Class Molecular Orbitals in Diatomics
 Theoretical Chemistry - Level II - Practical Class Molecular Orbitals in Diatomics Problem 1 Draw molecular orbital diagrams for O 2 and O 2 +. E / ev dioxygen molecule, O 2 dioxygenyl cation, O 2 + 25
Theoretical Chemistry - Level II - Practical Class Molecular Orbitals in Diatomics Problem 1 Draw molecular orbital diagrams for O 2 and O 2 +. E / ev dioxygen molecule, O 2 dioxygenyl cation, O 2 + 25
AN INTRODUCTION TO MOLECULAR ORBITALS
 AN INTRODUCTION TO MOLECULAR ORBITALS by YVES JEAN and FRANCOIS VOLATRON translated and edited by Jeremy Burdett New York Oxford OXFORD UNIVERSITY PRESS 1993 Contents Introduction, xiii I INTRODUCTION
AN INTRODUCTION TO MOLECULAR ORBITALS by YVES JEAN and FRANCOIS VOLATRON translated and edited by Jeremy Burdett New York Oxford OXFORD UNIVERSITY PRESS 1993 Contents Introduction, xiii I INTRODUCTION
Practical Issues on the Use of the CASPT2/CASSCF Method in Modeling Photochemistry: the Selection and Protection of an Active Space
 Practical Issues on the Use of the CASPT2/CASSCF Method in Modeling Photochemistry: the Selection and Protection of an Active Space Roland Lindh Dept. of Chemistry Ångström The Theoretical Chemistry Programme
Practical Issues on the Use of the CASPT2/CASSCF Method in Modeling Photochemistry: the Selection and Protection of an Active Space Roland Lindh Dept. of Chemistry Ångström The Theoretical Chemistry Programme
Conjugated Systems, Orbital Symmetry and UV Spectroscopy
 Conjugated Systems, Orbital Symmetry and UV Spectroscopy Introduction There are several possible arrangements for a molecule which contains two double bonds (diene): Isolated: (two or more single bonds
Conjugated Systems, Orbital Symmetry and UV Spectroscopy Introduction There are several possible arrangements for a molecule which contains two double bonds (diene): Isolated: (two or more single bonds
Multiconfigurational Quantum Chemistry. Björn O. Roos as told by RL Department of Theoretical Chemistry Chemical Center Lund University Sweden
 Multiconfigurational Quantum Chemistry Björn O. Roos as told by RL Department of Theoretical Chemistry Chemical Center Lund University Sweden April 20, 2009 1 The Slater determinant Using the spin-orbitals,
Multiconfigurational Quantum Chemistry Björn O. Roos as told by RL Department of Theoretical Chemistry Chemical Center Lund University Sweden April 20, 2009 1 The Slater determinant Using the spin-orbitals,
Q-Chem Workshop. Doubletree Hotel 2085 S. Harbor Boulevard Anaheim, CA March 26, Schedule
 Q-Chem Workshop Doubletree Hotel 2085 S. Harbor Boulevard Anaheim, CA 92802 March 26, 2011 1 8:30 Schedule Welcome remarks, Prof. Peter Gill, Australian National Univ and President of Q-Chem 8:45-9:15
Q-Chem Workshop Doubletree Hotel 2085 S. Harbor Boulevard Anaheim, CA 92802 March 26, 2011 1 8:30 Schedule Welcome remarks, Prof. Peter Gill, Australian National Univ and President of Q-Chem 8:45-9:15
Instructions for Using Spartan 14
 Instructions for Using Spartan 14 Log in to the computer with your Colby ID and password. Click on the Spartan 14 icon in the dock at the bottom of your screen. I. Building Molecules Spartan has one main
Instructions for Using Spartan 14 Log in to the computer with your Colby ID and password. Click on the Spartan 14 icon in the dock at the bottom of your screen. I. Building Molecules Spartan has one main
10.12 The Diels-Alder Reaction. Synthetic method for preparing compounds containing a cyclohexene ring
 10.12 The Diels-Alder Reaction Synthetic method for preparing compounds containing a cyclohexene ring In general... + conjugated diene alkene (dienophile) cyclohexene via transition state Mechanistic features
10.12 The Diels-Alder Reaction Synthetic method for preparing compounds containing a cyclohexene ring In general... + conjugated diene alkene (dienophile) cyclohexene via transition state Mechanistic features
CHEM 344 Molecular Modeling
 CHEM 344 Molecular Modeling The Use of Computational Chemistry to Support Experimental Organic Chemistry Day 1 all calculation data obtained from Gaussian09 using B3LYP/6-31G(d) unless otherwise noted.
CHEM 344 Molecular Modeling The Use of Computational Chemistry to Support Experimental Organic Chemistry Day 1 all calculation data obtained from Gaussian09 using B3LYP/6-31G(d) unless otherwise noted.
Nonadiabatic dynamics simulations of singlet fission in 2,5-bis(fluorene-9-ylidene)-2,5-dihydrothiophene crystals. Supporting Information.
-2,5-dihydrothiophene crystals. Supporting Information.") Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 18 Nonadiabatic dynamics simulations of singlet fission in,5-bis(fluorene-9-ylidene)-,5-dihydrothiophene
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 18 Nonadiabatic dynamics simulations of singlet fission in,5-bis(fluorene-9-ylidene)-,5-dihydrothiophene
SUPPLEMENTARY INFORMATION
 Calculations predict a stable molecular crystal of N 8 : Barak Hirshberg a, R. Benny Gerber a,b, and Anna I. Krylov c a Institute of Chemistry and The Fritz Haber Center for Molecular Dynamics, The Hebrew
Calculations predict a stable molecular crystal of N 8 : Barak Hirshberg a, R. Benny Gerber a,b, and Anna I. Krylov c a Institute of Chemistry and The Fritz Haber Center for Molecular Dynamics, The Hebrew
QUANTUM CHEMISTRY FOR TRANSITION METALS
 QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
and Ultraviolet Spectroscopy
 Organic Chemistry, 7 th Edition L. G. Wade, Jr. Chapter 15 Conjugated Systems, Orbital Symmetry, and Ultraviolet Spectroscopy 2010, Prentice all Conjugated Systems Conjugated double bonds are separated
Organic Chemistry, 7 th Edition L. G. Wade, Jr. Chapter 15 Conjugated Systems, Orbital Symmetry, and Ultraviolet Spectroscopy 2010, Prentice all Conjugated Systems Conjugated double bonds are separated
An Introduction to Quantum Chemistry and Potential Energy Surfaces. Benjamin G. Levine
 An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
NPA/NBO-Analysis. Examples POP =
 NPA/NBO-Analysis Examples POP = NBO Requests a full Natural Bond Orbital analysis, using NBO version 3 NPA Requests just the Natural Population Analysis phase of NBO. NBORead Requests a full NBO analysis,
NPA/NBO-Analysis Examples POP = NBO Requests a full Natural Bond Orbital analysis, using NBO version 3 NPA Requests just the Natural Population Analysis phase of NBO. NBORead Requests a full NBO analysis,
Basis Set for Molecular Orbital Theory
 Basis Set for Molecular Orbital Theory! Different Types of Basis Functions! Different Types of Atom Center Basis Functions! Classifications of Gaussian Basis Sets! Pseudopotentials! Molecular Properties
Basis Set for Molecular Orbital Theory! Different Types of Basis Functions! Different Types of Atom Center Basis Functions! Classifications of Gaussian Basis Sets! Pseudopotentials! Molecular Properties
QUANTUM CHEMISTRY PROJECT 3: ATOMIC AND MOLECULAR STRUCTURE
 Chemistry 460 Fall 2017 Dr. Jean M. Standard November 1, 2017 QUANTUM CHEMISTRY PROJECT 3: ATOMIC AND MOLECULAR STRUCTURE OUTLINE In this project, you will carry out quantum mechanical calculations of
Chemistry 460 Fall 2017 Dr. Jean M. Standard November 1, 2017 QUANTUM CHEMISTRY PROJECT 3: ATOMIC AND MOLECULAR STRUCTURE OUTLINE In this project, you will carry out quantum mechanical calculations of
QUANTUM CHEMISTRY WITH GAUSSIAN : A VERY BRIEF INTRODUCTION (PART 2)
") QUANTUM CHEMISTRY WITH GAUSSIAN : A VERY BRIEF INTRODUCTION (PART 2) TARAS V. POGORELOV AND MIKE HALLOCK SCHOOL OF CHEMICAL SCIENCES, UIUC This tutorial continues introduction to Gaussian [2]. Here we
QUANTUM CHEMISTRY WITH GAUSSIAN : A VERY BRIEF INTRODUCTION (PART 2) TARAS V. POGORELOV AND MIKE HALLOCK SCHOOL OF CHEMICAL SCIENCES, UIUC This tutorial continues introduction to Gaussian [2]. Here we
Chemistry 543--Final Exam--Keiderling May 5, pm SES
 Chemistry 543--Final Exam--Keiderling May 5,1992 -- 1-5pm -- 174 SES Please answer all questions in the answer book provided. Make sure your name is clearly indicated and that the answers are clearly numbered,
Chemistry 543--Final Exam--Keiderling May 5,1992 -- 1-5pm -- 174 SES Please answer all questions in the answer book provided. Make sure your name is clearly indicated and that the answers are clearly numbered,
Gaussian: Basic Tutorial
 Input file: # hf sto-g pop=full Water - Single Point Energy 0 H.0 H.0 H 04.5 Route Section Start with # Contains the keywords Gaussian: Basic Tutorial Route Section Title Section Charge-Multiplicity Molecule
Input file: # hf sto-g pop=full Water - Single Point Energy 0 H.0 H.0 H 04.5 Route Section Start with # Contains the keywords Gaussian: Basic Tutorial Route Section Title Section Charge-Multiplicity Molecule
The MCSCF Method *, Molecular Orbitals, Reference Spaces and COLUMBUS Input
 The MCSCF Method *, Molecular Orbitals, Reference Spaces and COLUMBUS Input Hans Lischka University of Vienna *Excerpt of a course presented by R. Shepard, Argonne National Laboratory, at the Workshop
The MCSCF Method *, Molecular Orbitals, Reference Spaces and COLUMBUS Input Hans Lischka University of Vienna *Excerpt of a course presented by R. Shepard, Argonne National Laboratory, at the Workshop
ACES2 Labs. Part B: Calculating electronically excited, ionized, and electro-attached states using equation of motion coupled cluster calculations.
 ACES2 Labs. Part B: Calculating electronically excited, ionized, and electro-attached states using equation of motion coupled cluster calculations. I. Electronically excited states. Purpose: - Calculation
ACES2 Labs. Part B: Calculating electronically excited, ionized, and electro-attached states using equation of motion coupled cluster calculations. I. Electronically excited states. Purpose: - Calculation
Lab #3: Choice of Theoretical Method
 Lab #3: Choice of Theoretical Method These directions assume the user is familiar with the WebMO interface and can build molecules, set up calculations, etc. Exercise 1 - Determine the Proton Affinity
Lab #3: Choice of Theoretical Method These directions assume the user is familiar with the WebMO interface and can build molecules, set up calculations, etc. Exercise 1 - Determine the Proton Affinity
Chemistry 5021/8021 Computational Chemistry 3/4 Credits Spring Semester 2004 ( Due 4 / 5 / 04 )
") Chemistry 5021/8021 Computational Chemistry 3/4 Credits Spring Semester 2004 ( Due 4 / 5 / 04 ) This problem set will take longer than the last one in the sense that you may need to submit some jobs, leave,
Chemistry 5021/8021 Computational Chemistry 3/4 Credits Spring Semester 2004 ( Due 4 / 5 / 04 ) This problem set will take longer than the last one in the sense that you may need to submit some jobs, leave,
Introduction to Computational Chemistry for Experimental Chemists... (Part 2/2)
") 12 th PhD seminar, Garching, October 31 st 2008 Introduction to Computational Chemistry for Experimental Chemists... (Part 2/2) Dr. Markus Drees, TU München Universität Regensburg Universität Augsburg
12 th PhD seminar, Garching, October 31 st 2008 Introduction to Computational Chemistry for Experimental Chemists... (Part 2/2) Dr. Markus Drees, TU München Universität Regensburg Universität Augsburg
Excited States Calculations for Protonated PAHs
 52 Chapter 3 Excited States Calculations for Protonated PAHs 3.1 Introduction Protonated PAHs are closed shell ions. Their electronic structure should therefore be similar to that of neutral PAHs, but
52 Chapter 3 Excited States Calculations for Protonated PAHs 3.1 Introduction Protonated PAHs are closed shell ions. Their electronic structure should therefore be similar to that of neutral PAHs, but
AB INITIO MODELING OF THE STRUCTURAL DEFECTS IN AMIDES
 Int. J. Chem. Sci.: 9(4), 2011, 1564-1568 ISSN 0972-768X www.sadgurupublications.com AB INITIO MODELING OF THE STRUCTURAL DEFECTS IN AMIDES M. FATHIMA BEGUM, HEMA TRESA VARGHESE a, Y. SHEENA MARY a, C.
Int. J. Chem. Sci.: 9(4), 2011, 1564-1568 ISSN 0972-768X www.sadgurupublications.com AB INITIO MODELING OF THE STRUCTURAL DEFECTS IN AMIDES M. FATHIMA BEGUM, HEMA TRESA VARGHESE a, Y. SHEENA MARY a, C.
7 Infrared, Thermochemistry, UV-Vis, and NMR
 7 Infrared, Thermochemistry, UV-Vis, and NMR Exercise 1 Method Dependence and Scaling for the Infrared Spectrum of Formaldehyde. Build a molecule of formaldehyde using sp 2 C and atoms. Clean up the structure
7 Infrared, Thermochemistry, UV-Vis, and NMR Exercise 1 Method Dependence and Scaling for the Infrared Spectrum of Formaldehyde. Build a molecule of formaldehyde using sp 2 C and atoms. Clean up the structure
This is called a singlet or spin singlet, because the so called multiplicity, or number of possible orientations of the total spin, which is
 9. Open shell systems The derivation of Hartree-Fock equations (Chapter 7) was done for a special case of a closed shell systems. Closed shell means that each MO is occupied by two electrons with the opposite
9. Open shell systems The derivation of Hartree-Fock equations (Chapter 7) was done for a special case of a closed shell systems. Closed shell means that each MO is occupied by two electrons with the opposite
NH 3 H 2 O N 2. Why do they make chemical bonds? Molecular Orbitals
 N 2 NH 3 H 2 O Why do they make chemical bonds? 5 Molecular Orbitals Why do they make chemical bonds? Stabilization Bond energy Types of Chemical Bonds Metallic Bond Ionic Bond Covalent Bond Covalent Bond
N 2 NH 3 H 2 O Why do they make chemical bonds? 5 Molecular Orbitals Why do they make chemical bonds? Stabilization Bond energy Types of Chemical Bonds Metallic Bond Ionic Bond Covalent Bond Covalent Bond
NWChem: Coupled Cluster Method (Tensor Contraction Engine)
") NWChem: Coupled Cluster Method (Tensor Contraction Engine) Why CC is important?! Correlation effects are important!! CC is size-extensive theory: can be used to describe dissociation processes.! Higher-order
NWChem: Coupled Cluster Method (Tensor Contraction Engine) Why CC is important?! Correlation effects are important!! CC is size-extensive theory: can be used to describe dissociation processes.! Higher-order
PART II ADVANCED AND SPECIAL SUBJECTS
 PART II ADVANCED AND SPECIAL SUBJECTS 1 PART II ADVANCED AND SPECIAL SUBJECTS PART II ADVANCED AND SPECIAL SUBJECTS 2 1 Computer Experiment 7: Interpretation of Structure, Bonding and Reactivity Using
PART II ADVANCED AND SPECIAL SUBJECTS 1 PART II ADVANCED AND SPECIAL SUBJECTS PART II ADVANCED AND SPECIAL SUBJECTS 2 1 Computer Experiment 7: Interpretation of Structure, Bonding and Reactivity Using
Organic Chemistry: CHEM2322
 Conjugated Systems Organic Chemistry: We met in Chem 2321 unsaturated bonds as either a C=C bond or C C bond. If these unsaturated bonds are well separated then they react independently however if there
Conjugated Systems Organic Chemistry: We met in Chem 2321 unsaturated bonds as either a C=C bond or C C bond. If these unsaturated bonds are well separated then they react independently however if there
5.4. Electronic structure of water
 5.4. Electronic structure of water Water belongs to C 2v point group, we have discussed the corresponding character table. Here it is again: C 2v E C 2 σ v (yz) σ v (xz) A 1 1 1 1 1 A 2 1 1-1 -1 B 1 1-1
5.4. Electronic structure of water Water belongs to C 2v point group, we have discussed the corresponding character table. Here it is again: C 2v E C 2 σ v (yz) σ v (xz) A 1 1 1 1 1 A 2 1 1-1 -1 B 1 1-1
Applications of Computational Chemistry to the Reactions of Lignin
 Applications of Computational Chemistry to the Reactions of Lignin Thomas Elder USDA-Forest Service Southern Research Station ctober 30-ovember 2, 2012 Frontiers in Biorefining, Chemicals and Products
Applications of Computational Chemistry to the Reactions of Lignin Thomas Elder USDA-Forest Service Southern Research Station ctober 30-ovember 2, 2012 Frontiers in Biorefining, Chemicals and Products
Exercise 1: Structure and dipole moment of a small molecule
 Introduction to computational chemistry Exercise 1: Structure and dipole moment of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the dipole moment of a small
Introduction to computational chemistry Exercise 1: Structure and dipole moment of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the dipole moment of a small
Orbital Density Dependent Functionals
 Orbital Density Dependent Functionals S. Kluepfel1, P. Kluepfel1, Hildur Guðmundsdóttir1 and Hannes Jónsson1,2 1. Univ. of Iceland; 2. Aalto University Outline: Problems with GGA approximation (PBE, RPBE,...)
Orbital Density Dependent Functionals S. Kluepfel1, P. Kluepfel1, Hildur Guðmundsdóttir1 and Hannes Jónsson1,2 1. Univ. of Iceland; 2. Aalto University Outline: Problems with GGA approximation (PBE, RPBE,...)
Structural and Theoretical Studies of 2-amino-3- nitropyridine
 ISSN: 0973-4945; CODEN ECJHAO E-Journal of Chemistry http://www.ejchem.net 2012, 9(4), 2191-2204 Structural and Theoretical Studies of 2-amino-3- nitropyridine N. S. AL-HOKBANY a, A. A. DAHY b, I. KH.WARAD
ISSN: 0973-4945; CODEN ECJHAO E-Journal of Chemistry http://www.ejchem.net 2012, 9(4), 2191-2204 Structural and Theoretical Studies of 2-amino-3- nitropyridine N. S. AL-HOKBANY a, A. A. DAHY b, I. KH.WARAD
Learning to Use Scigress Wagner, Eugene P. (revised May 15, 2018)
") Learning to Use Scigress Wagner, Eugene P. (revised May 15, 2018) Abstract Students are introduced to basic features of Scigress by building molecules and performing calculations on them using semi-empirical
Learning to Use Scigress Wagner, Eugene P. (revised May 15, 2018) Abstract Students are introduced to basic features of Scigress by building molecules and performing calculations on them using semi-empirical
2018 Ch112 problem set 6 Due: Thursday, Dec. 6th. Problem 1 (2 points)
") Problem 1 (2 points) a. Consider the following V III complexes: V(H2O)6 3+, VF6 3-, and VCl6 3-. The table below contains the energies corresponding to the two lowest spin-allowed d-d transitions (υ1 and
Problem 1 (2 points) a. Consider the following V III complexes: V(H2O)6 3+, VF6 3-, and VCl6 3-. The table below contains the energies corresponding to the two lowest spin-allowed d-d transitions (υ1 and
General Chemistry I (2012) Lecture by B. H. Hong
 Lecture by B. H. Hong") 3.8 The Limitations of Lewis's Theory 3.9 Molecular Orbitals The valence-bond (VB) and molecular orbital (MO) theories are both procedures for constructing approximate wavefunctions of electrons. The MO
3.8 The Limitations of Lewis's Theory 3.9 Molecular Orbitals The valence-bond (VB) and molecular orbital (MO) theories are both procedures for constructing approximate wavefunctions of electrons. The MO
Lecture B6 Molecular Orbital Theory. Sometimes it's good to be alone.
 Lecture B6 Molecular Orbital Theory Sometimes it's good to be alone. Covalent Bond Theories 1. VSEPR (valence shell electron pair repulsion model). A set of empirical rules for predicting a molecular geometry
Lecture B6 Molecular Orbital Theory Sometimes it's good to be alone. Covalent Bond Theories 1. VSEPR (valence shell electron pair repulsion model). A set of empirical rules for predicting a molecular geometry
The successful wavefunction can be written as a determinant: # 1 (2) # 2 (2) Electrons. This can be generalized to our 2N-electron wavefunction:
 # 2 (2) Electrons. This can be generalized to our 2N-electron wavefunction:") T2. CNDO to AM1: The Semiempirical Molecular Orbital Models The discussion in sections T2.1 T2.3 applies also to ab initio molecular orbital calculations. T2.1 Slater Determinants Consider the general
T2. CNDO to AM1: The Semiempirical Molecular Orbital Models The discussion in sections T2.1 T2.3 applies also to ab initio molecular orbital calculations. T2.1 Slater Determinants Consider the general
Bonds in molecules are formed by the interactions between electrons.
 CHEM 2060 Lecture 6: Electrostatic Interactions L6-1 PART TWO: Electrostatic Interactions In the first section of this course, we were more concerned with structural aspects of molecules. In this section
CHEM 2060 Lecture 6: Electrostatic Interactions L6-1 PART TWO: Electrostatic Interactions In the first section of this course, we were more concerned with structural aspects of molecules. In this section
Bonding/Lewis Dots Lecture Page 1 of 12 Date. Bonding. What is Coulomb's Law? Energy Profile: Covalent Bonds. Electronegativity and Linus Pauling
 Bonding/Lewis Dots Lecture Page 1 of 12 Date Bonding What is Coulomb's Law? Energy Profile: Covalent Bonds Electronegativity and Linus Pauling 2.1 H 1.0 Li 0.9 Na 0.8 K 0.8 Rb 0.7 Cs 0.7 Fr 1.5 Be 1.2
Bonding/Lewis Dots Lecture Page 1 of 12 Date Bonding What is Coulomb's Law? Energy Profile: Covalent Bonds Electronegativity and Linus Pauling 2.1 H 1.0 Li 0.9 Na 0.8 K 0.8 Rb 0.7 Cs 0.7 Fr 1.5 Be 1.2
CHEM1102 Worksheet 4 Answers to Critical Thinking Questions Model 1: Infrared (IR) Spectroscopy
 Spectroscopy") CEM1102 Worksheet 4 Answers to Critical Thinking Questions Model 1: Infrared (IR) Spectroscopy 1. See below. Model 2: UV-Visible Spectroscopy 1. See below. 2. All of the above. 3. Restricted to the identification
CEM1102 Worksheet 4 Answers to Critical Thinking Questions Model 1: Infrared (IR) Spectroscopy 1. See below. Model 2: UV-Visible Spectroscopy 1. See below. 2. All of the above. 3. Restricted to the identification