Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design
|
|
|
- Imogene Peters
- 5 years ago
- Views:
Transcription
1 Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1


2 Computer Aided Drug Design - Introduction Development of computers - hardware - algorithms softwares - computation chemists - representation 2
3 Computer Aided Drug Design - Introduction Models Molecular modelling methods - Molecular or quantum mechanics - Semiempirical vs ab initio methods - Cartesian and polar coordinates 3




4 Computer Aided Drug Design - Introduction Computer graphics - space fill, CPK (Corey-Pauling-Koulton), stick, stick and ball, mesh, ribbon, surface, molecular dinamics etc. 4
5 Molecular Mechanics Basis: E total = Σ attractive + repulsive forces - mechanical method atoms are balls with respective atomic masses 5
6 Molecular Mechanics E total = Σ E stretching +Σ E bend +ΣE torsion +ΣE vdw +ΣE coulombic 6
7 Molecular Mechanics Creating a molecular model using molecular mechanics - Joining fragments from program database - Prepare 2D structure and convert it to 3D structure - Converting an existing model from the database 7

8 Molecular Dynamics Creating a dynamic molecular model - atomic coordinates (twist, bend, stretch etc.) - conformational analysis 8
 ab initio")

9 Quantum Mechanics Schrodinger equation, Born-Oppenheimer approximation - simplification Hartree-Fock approximation Density Funtional Theory Practice Gaussian 03 (or 98) ab initio methods Gamess The Nobel Prize in Chemistry 1998 Walter Kohn "for his development of the density-functional theory" John A. Pople "for his development of computational methods in quantum chemistry" 9

 (ligand) E")

10 Docking Produce and investigate the complex of large biomolecule (host) and the drug (or candidate) (ligand) E binding = E target + E ligand E target-ligand Global E min Bioactive conformer 10
11 Docking De novo design Using docking programs to design new lead structures The template method The component fragment method 11
12 Docking A case study Inhibition of FBPase (Kimberly Stieglitz) Start: - build and optimize the drug candidate ChemDraw and import to Gaussian or get crystal structure (CCD) - get protein (enzyme or receptor) structure PDB 12
13 Docking A case study Inhibition of FBPase - list of compounds 13


14 Docking A case study Inhibition of FBPase - protein structure 14
15 Docking A case study Inhibition of FBPase - docking 15

16 16


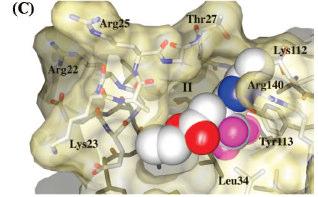

17 Docking A case study Inhibition of FBPase - visualization 17
18 Docking 18
19 Docking 19
20 Docking 20

21 Comparing 3D Structure by Overlays 21

22 Comparing 3D Structure by Overlays 22

23 23


24 Pharmacophores A case study Inhibition of AChE - Marianna Torok, Seema Bag 24
25 Pharmacophores Typical pharmacophore features are: - Hydrophobic - Aromatic - Hydrogen bond donor/acceptor - Cation/anion The features need to match different chemical groups with similar properties, in order to identify novel ligands. Ligands receptor interactions are typically polar positive, polar negative or hydrophobic. A well-defined pharmacophore model includes both hydrophobic volumes and hydrogen bond vectors. 25
26 Pharmacophores Select molecules: highly active molecules Training set + test set Prepare Ligands PHASE Version 3.0 Find common pharmacophore using binary decision tree Create pharmacophoric sites Alignment Build QSAR model r 2 & q2 value Screen/design for more potent analogs 26

27 Pharmacophores Pharmacophoric points on active molecules 27

28 Interpretation Pharmacophores H-donor Hydrophobic 28
29 Pharmacophores 29

30 Pharmacophores galanthamine dihydrocodein morphine 30

31 31
32 Modeling Protein Structures Solvent Mapping (Sandor Vajda, BU) 16 solvents: ethanol, acetaldehyde, acetonitrile, benzaldehyde, isopropanol, methanol, dimethyl ether, urea, benzene, t-butanol, isobutanol, cyclohexane, methanamine, acetamide, phenol, acetone, dimethylformamide and ethane) will be used in rigid-body fragment docking 32


33 Modeling Protein Structures 33

34 34
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
5.1. Hardwares, Softwares and Web server used in Molecular modeling
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit
 October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Medicinal Chemistry/ CHEM 458/658 Chapter 3- SAR and QSAR
 Medicinal Chemistry/ CHEM 458/658 Chapter 3- SAR and QSAR Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Introduction Structure-Activity Relationship (SAR) - similar
Medicinal Chemistry/ CHEM 458/658 Chapter 3- SAR and QSAR Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Introduction Structure-Activity Relationship (SAR) - similar
Supplementary Information for Evaluating the. energetic driving force for co-crystal formation
 Supplementary Information for Evaluating the energetic driving force for co-crystal formation Christopher R. Taylor and Graeme M. Day School of Chemistry, University of Southampton, Highfield, Southampton,
Supplementary Information for Evaluating the energetic driving force for co-crystal formation Christopher R. Taylor and Graeme M. Day School of Chemistry, University of Southampton, Highfield, Southampton,
CHEM 545 Theory and Practice of Molecular Electronic Structure. Anna I. Krylov. DON T PANIC.
 CHEM 545 Theory and Practice of Molecular Electronic Structure Anna I. Krylov http://iopenshell.usc.edu/chem545/ DON T PANIC USC Fall 2014 Things to do: 1. Install IQmol (by this Thursday). http://iqmol.org/.
CHEM 545 Theory and Practice of Molecular Electronic Structure Anna I. Krylov http://iopenshell.usc.edu/chem545/ DON T PANIC USC Fall 2014 Things to do: 1. Install IQmol (by this Thursday). http://iqmol.org/.
COMPUTERS IN PHARMACEUTICAL RESEARCH & DEVELOPMENT
 COMPUTERS IN PHARMACEUTICAL RESEARCH & DEVELOPMENT Presented by MrsA. Lavanya M.Pharm., Assistant Professor Department of Pharmaceutics Krishna Teja Pharmacy college Subject; Computer Aided Drug Delivery
COMPUTERS IN PHARMACEUTICAL RESEARCH & DEVELOPMENT Presented by MrsA. Lavanya M.Pharm., Assistant Professor Department of Pharmaceutics Krishna Teja Pharmacy college Subject; Computer Aided Drug Delivery
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
CSD. CSD-Enterprise. Access the CSD and ALL CCDC application software
 CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining
![Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining](/thumbs/91/107031261.jpg "Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining") Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics
 Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites. J. Andrew Surface
 Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
The Conformation Search Problem
 Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Introduction to Chemoinformatics and Drug Discovery
 Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
MODELING AND INFORMATICS IN DRUG DESIGN. National Institute of Pharmaceutical Education and Research (NIPER), S.A.S. Nagar, India
, S.A.S. Nagar, India") 1 MODELING AND INFORMATICS IN DRUG DESIGN Prasad V. Bharatam,* Smriti Khanna, and Sandrea M. Francis National Institute of Pharmaceutical Education and Research (NIPER), S.A.S. Nagar, India Contents 1.1
1 MODELING AND INFORMATICS IN DRUG DESIGN Prasad V. Bharatam,* Smriti Khanna, and Sandrea M. Francis National Institute of Pharmaceutical Education and Research (NIPER), S.A.S. Nagar, India Contents 1.1
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Additional background material on the Nobel Prize in Chemistry 1998
 Additional background material on the Nobel Prize in Chemistry 1998 The Royal Swedish Academy of Sciences has decided to award the 1998 Nobel Prize in Chemistry with one half to Professor WALTER KOHN,
Additional background material on the Nobel Prize in Chemistry 1998 The Royal Swedish Academy of Sciences has decided to award the 1998 Nobel Prize in Chemistry with one half to Professor WALTER KOHN,
Identifying Interaction Hot Spots with SuperStar
 Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Schrodinger ebootcamp #3, Summer EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist
 Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Nonlinear QSAR and 3D QSAR
 onlinear QSAR and 3D QSAR Hugo Kubinyi Germany E-Mail kubinyi@t-online.de HomePage www.kubinyi.de onlinear Lipophilicity-Activity Relationships drug receptor Possible Reasons for onlinear Lipophilicity-Activity
onlinear QSAR and 3D QSAR Hugo Kubinyi Germany E-Mail kubinyi@t-online.de HomePage www.kubinyi.de onlinear Lipophilicity-Activity Relationships drug receptor Possible Reasons for onlinear Lipophilicity-Activity
Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations
 Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations Alexandre V. Morozov, Tanja Kortemme, Kiril Tsemekhman, David Baker
Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations Alexandre V. Morozov, Tanja Kortemme, Kiril Tsemekhman, David Baker
Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,..
 3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Computational analysis of the activity of pongachalcone I against highly resistant bacteria Pseudomonas putida
 Computational analysis of the activity of pongachalcone I against highly resistant bacteria Pseudomonas putida Satya B. Paul 1, Sudip Choudhury 2,* 1 Department of Chemistry, Assam University, Silchar,
Computational analysis of the activity of pongachalcone I against highly resistant bacteria Pseudomonas putida Satya B. Paul 1, Sudip Choudhury 2,* 1 Department of Chemistry, Assam University, Silchar,
Computer Graphics Applications on Molecular Biology and Drug Design
 Computer Graphics Applications on Molecular Biology and Drug Design Katerina PERDIKURI 1, Athanasios TSAKALIDIS 1 1 Department of Computer Engineering and Informatics University of Patras,26500 Patras,
Computer Graphics Applications on Molecular Biology and Drug Design Katerina PERDIKURI 1, Athanasios TSAKALIDIS 1 1 Department of Computer Engineering and Informatics University of Patras,26500 Patras,
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Molecular Modelling. Computational Chemistry Demystified. RSC Publishing. Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK
 Molecular Modelling Computational Chemistry Demystified Peter Bladon Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK John E. Gorton Gorton Systems, Glasgow, UK Robert B. Hammond Institute
Molecular Modelling Computational Chemistry Demystified Peter Bladon Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK John E. Gorton Gorton Systems, Glasgow, UK Robert B. Hammond Institute
Drug Design 2. Oliver Kohlbacher. Winter 2009/ QSAR Part 4: Selected Chapters
 Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
What is Protein-Ligand Docking?
 MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Hit Finding and Optimization Using BLAZE & FORGE
 Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Ferdowsi University of Mashhad
 Spectroscopy in Inorganic Chemistry 2 Diatomic molecule C v and D h HCN H-H 3 contribution orbital electron Σ 0 σ 1 Π 1 π 1 Δ 2 δ 1 Φ 3 δ 1 Σ + Σ - 4 Linear molecule NO 2s+1 2 Π A 1 =Σ + 0 A 2 =Σ - 0 E
Spectroscopy in Inorganic Chemistry 2 Diatomic molecule C v and D h HCN H-H 3 contribution orbital electron Σ 0 σ 1 Π 1 π 1 Δ 2 δ 1 Φ 3 δ 1 Σ + Σ - 4 Linear molecule NO 2s+1 2 Π A 1 =Σ + 0 A 2 =Σ - 0 E
Toward an Understanding of GPCR-ligand Interactions. Alexander Heifetz
 Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
6.02 Molecular Modelling (5 points)
") 2nd/3rd Year Physical Chemistry Practical Course, Oxford University 6.02 Molecular Modelling (5 points) Introduction The need for computer modelling Traditionally chemists have synthesized molecules and
2nd/3rd Year Physical Chemistry Practical Course, Oxford University 6.02 Molecular Modelling (5 points) Introduction The need for computer modelling Traditionally chemists have synthesized molecules and
Spectroscopy in Inorganic Chemistry. Electronic Absorption Spectroscopy
 Spectroscopy in Inorganic Chemistry Diatomic molecule C v and D h NO H-H 2 contribution orbital Σ 0 σ Π 1 π Δ 2 δ Φ 3 δ 3 Linear molecule NO 2s+1 2 Π A 1 =Σ + 0 A 2 =Σ - 0 E 1 =Π 1 E 2 =Δ 2 E 3 =Φ 3 4
Spectroscopy in Inorganic Chemistry Diatomic molecule C v and D h NO H-H 2 contribution orbital Σ 0 σ Π 1 π Δ 2 δ Φ 3 δ 3 Linear molecule NO 2s+1 2 Π A 1 =Σ + 0 A 2 =Σ - 0 E 1 =Π 1 E 2 =Δ 2 E 3 =Φ 3 4
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Medicinal Chemistry/ CHEM 458/658 Chapter 8- Receptors and Messengers
 Medicinal Chemistry/ CHEM 458/658 Chapter 8- Receptors and Messengers Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Introduction Receptor specific areas of proteins
Medicinal Chemistry/ CHEM 458/658 Chapter 8- Receptors and Messengers Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Introduction Receptor specific areas of proteins
Chem 1140; Molecular Modeling
 P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
The mapping of a protein by experimental or computational tools
 Computational mapping identifies the binding sites of organic solvents on proteins Sheldon Dennis, Tamas Kortvelyesi, and Sandor Vajda Department of Biomedical Engineering, Boston University, Boston, MA
Computational mapping identifies the binding sites of organic solvents on proteins Sheldon Dennis, Tamas Kortvelyesi, and Sandor Vajda Department of Biomedical Engineering, Boston University, Boston, MA
Chemistry 343- Spring 2008
 Chemistry 343- Spring 2008 27 Chapter 2- Representative Carbon Compounds: Functional Groups, Intermolecular Forces and IR Spectroscopy A. ydrocarbons: Compounds composed of only C and Four Basic Types:
Chemistry 343- Spring 2008 27 Chapter 2- Representative Carbon Compounds: Functional Groups, Intermolecular Forces and IR Spectroscopy A. ydrocarbons: Compounds composed of only C and Four Basic Types:
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy
 Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
ICM-Chemist-Pro How-To Guide. Version 3.6-1h Last Updated 12/29/2009
 ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
Aromatic Hydrocarbons
 Aromatic Hydrocarbons Aromatic hydrocarbons contain six-membered rings of carbon atoms with alternating single and double carbon-carbon bonds. The ring is sometimes shown with a circle in the center instead
Aromatic Hydrocarbons Aromatic hydrocarbons contain six-membered rings of carbon atoms with alternating single and double carbon-carbon bonds. The ring is sometimes shown with a circle in the center instead
Molecular Modelling for Medicinal Chemistry (F13MMM) Room A36
 Room A36") Molecular Modelling for Medicinal Chemistry (F13MMM) jonathan.hirst@nottingham.ac.uk Room A36 http://comp.chem.nottingham.ac.uk Assisted reading Molecular Modelling: Principles and Applications. Andrew
Molecular Modelling for Medicinal Chemistry (F13MMM) jonathan.hirst@nottingham.ac.uk Room A36 http://comp.chem.nottingham.ac.uk Assisted reading Molecular Modelling: Principles and Applications. Andrew
Molecular Visualization. Introduction
 Molecular Visualization Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences Duquesne University Introduction Assessments of change, dynamics, and cause and effect
Molecular Visualization Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences Duquesne University Introduction Assessments of change, dynamics, and cause and effect
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
SCULPT 3.0. Using SCULPT to Gain Competitive Insights. Brings 3D Visualization to the Lab Bench SPECIAL REPORT. 4 Molecular Connection Fall 1999
 SPECIAL REPORT SCULPT 3.0 Brings 3D Visualization to the Lab Bench ith the acquisition of Interactive Simulations, Inc., last April, MDL added to its arsenal of discovery informatics tools a powerful analysis
SPECIAL REPORT SCULPT 3.0 Brings 3D Visualization to the Lab Bench ith the acquisition of Interactive Simulations, Inc., last April, MDL added to its arsenal of discovery informatics tools a powerful analysis
Different conformations of the drugs within the virtual library of FDA approved drugs will be generated.
 Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
Ping-Chiang Lyu. Institute of Bioinformatics and Structural Biology, Department of Life Science, National Tsing Hua University.
 Pharmacophore-based Drug design Ping-Chiang Lyu Institute of Bioinformatics and Structural Biology, Department of Life Science, National Tsing Hua University 96/08/07 Outline Part I: Analysis The analytical
Pharmacophore-based Drug design Ping-Chiang Lyu Institute of Bioinformatics and Structural Biology, Department of Life Science, National Tsing Hua University 96/08/07 Outline Part I: Analysis The analytical
Solutions and Non-Covalent Binding Forces
 Chapter 3 Solutions and Non-Covalent Binding Forces 3.1 Solvent and solution properties Molecules stick together using the following forces: dipole-dipole, dipole-induced dipole, hydrogen bond, van der
Chapter 3 Solutions and Non-Covalent Binding Forces 3.1 Solvent and solution properties Molecules stick together using the following forces: dipole-dipole, dipole-induced dipole, hydrogen bond, van der
Computational Modeling of Protein-Ligand Interactions
 Computational Modeling of Protein-Ligand Interactions Steven R. Gwaltney Department of Chemistry Mississippi State University Mississippi State, MS 39762 Auguste Comte, 1830 Every attempt to refer chemical
Computational Modeling of Protein-Ligand Interactions Steven R. Gwaltney Department of Chemistry Mississippi State University Mississippi State, MS 39762 Auguste Comte, 1830 Every attempt to refer chemical
Ligand-receptor interactions
 University of Silesia, Katowice, Poland 11 22 March 2013 Ligand-receptor interactions Dr. Pavel Polishchuk A.V. Bogatsky Physico-Chemical Institute of National Academy of Sciences of Ukraine Odessa, Ukraine
University of Silesia, Katowice, Poland 11 22 March 2013 Ligand-receptor interactions Dr. Pavel Polishchuk A.V. Bogatsky Physico-Chemical Institute of National Academy of Sciences of Ukraine Odessa, Ukraine
Advanced in silico drug design
 Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
More information can be found in Chapter 12 in your textbook for CHEM 3750/ 3770 and on pages in your laboratory manual.
 CHEM 3780 rganic Chemistry II Infrared Spectroscopy and Mass Spectrometry Review More information can be found in Chapter 12 in your textbook for CHEM 3750/ 3770 and on pages 13-28 in your laboratory manual.
CHEM 3780 rganic Chemistry II Infrared Spectroscopy and Mass Spectrometry Review More information can be found in Chapter 12 in your textbook for CHEM 3750/ 3770 and on pages 13-28 in your laboratory manual.
Molecular modeling with InsightII
 Molecular modeling with InsightII Yuk Sham Computational Biology/Biochemistry Consultant Phone: (612) 624 7427 (Walter Library) Phone: (612) 624 0783 (VWL) Email: shamy@msi.umn.edu How to run InsightII
Molecular modeling with InsightII Yuk Sham Computational Biology/Biochemistry Consultant Phone: (612) 624 7427 (Walter Library) Phone: (612) 624 0783 (VWL) Email: shamy@msi.umn.edu How to run InsightII
Computational Solvent Mapping Reveals the Importance of Local Conformational Changes for Broad Substrate Specificity in Mammalian Cytochromes P450
 Biochemistry 2006, 45, 9393-9407 9393 Computational Solvent Mapping Reveals the Importance of Local Conformational Changes for Broad Substrate Specificity in Mammalian Cytochromes P450 Karl H. Clodfelter,
Biochemistry 2006, 45, 9393-9407 9393 Computational Solvent Mapping Reveals the Importance of Local Conformational Changes for Broad Substrate Specificity in Mammalian Cytochromes P450 Karl H. Clodfelter,
Computational Material Science Part II
 Computational Material Science Part II Ito Chao ( ) Institute of Chemistry Academia Sinica Aim of Part II Get familiar with the computational methodologies often used and properties often predicted in
Computational Material Science Part II Ito Chao ( ) Institute of Chemistry Academia Sinica Aim of Part II Get familiar with the computational methodologies often used and properties often predicted in
Introduction to Hartree-Fock Molecular Orbital Theory
 Introduction to Hartree-Fock Molecular Orbital Theory C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Origins of Mathematical Modeling in Chemistry Plato (ca. 428-347
Introduction to Hartree-Fock Molecular Orbital Theory C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Origins of Mathematical Modeling in Chemistry Plato (ca. 428-347
Navigation in Chemical Space Towards Biological Activity. Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland
 Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Cres s et Bi omol ecul a r Di s covery Ltd, a l l ri ghts res erved.
 10.5 2017 Cres s et Bi omol ecul a r Di s covery Ltd, a l l ri ghts res erved. Table of contents Introduction... 8 What are field points?... 8 Interpretation of field point patterns... 9 About this document...
10.5 2017 Cres s et Bi omol ecul a r Di s covery Ltd, a l l ri ghts res erved. Table of contents Introduction... 8 What are field points?... 8 Interpretation of field point patterns... 9 About this document...
Quantum Chemical Simulations and Descriptors. Dr. Antonio Chana, Dr. Mosè Casalegno
 Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Dock Ligands from a 2D Molecule Sketch
 Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
Principles of Drug Design
 Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction
Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction
Data Quality Issues That Can Impact Drug Discovery
 Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Performing a Pharmacophore Search using CSD-CrossMiner
 Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
IFM Chemistry Computational Chemistry 2010, 7.5 hp LAB2. Computer laboratory exercise 1 (LAB2): Quantum chemical calculations
: Quantum chemical calculations") Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Targeting protein-protein interactions: A hot topic in drug discovery
 Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
ENERGY MINIMIZATION AND CONFORMATION SEARCH ANALYSIS OF TYPE-2 ANTI-DIABETES DRUGS
 Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses
 Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
COMPUTER AIDED DRUG DESIGN (CADD) AND DEVELOPMENT METHODS
 AND DEVELOPMENT METHODS") COMPUTER AIDED DRUG DESIGN (CADD) AND DEVELOPMENT METHODS DRUG DEVELOPMENT Drug development is a challenging path Today, the causes of many diseases (rheumatoid arthritis, cancer, mental diseases, etc.)
COMPUTER AIDED DRUG DESIGN (CADD) AND DEVELOPMENT METHODS DRUG DEVELOPMENT Drug development is a challenging path Today, the causes of many diseases (rheumatoid arthritis, cancer, mental diseases, etc.)
Molecular Modeling Study of Some Anthelmintic 2-phenyl Benzimidazole-1- Acetamides as β-tubulin Inhibitor
 Sawant et al : Molecular Modeling Study of Some Anthelmintic 2-phenyl Benzimidazole-1-Acetamides as -tubulin Inhibitor 1269 International Journal of Drug Design and Discovery Volume 5 Issue 1 January March
Sawant et al : Molecular Modeling Study of Some Anthelmintic 2-phenyl Benzimidazole-1-Acetamides as -tubulin Inhibitor 1269 International Journal of Drug Design and Discovery Volume 5 Issue 1 January March
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification
 Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Paper No. 1: ORGANIC CHEMISTRY- I (Nature of Bonding and Stereochemistry)
") Subject Chemistry Paper No and Title Paper 1: ORGANIC - I (Nature of Bonding Module No and Title Module Tag CHE_P1_M10 TABLE OF CONTENTS 1. Learning Outcomes 2. Introduction 3. Non-Covalent Interactions
Subject Chemistry Paper No and Title Paper 1: ORGANIC - I (Nature of Bonding Module No and Title Module Tag CHE_P1_M10 TABLE OF CONTENTS 1. Learning Outcomes 2. Introduction 3. Non-Covalent Interactions
Integrated Cheminformatics to Guide Drug Discovery
 Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Molecules Are NOT Static Structures!
 Objective 6 Draw conformational isomers of chains (staggered, eclipsed) and rings (chair axial/equatorial, boat, cis/trans) using skeletal structures, Newman projections, wedge-dash, sawhorse. Identify
Objective 6 Draw conformational isomers of chains (staggered, eclipsed) and rings (chair axial/equatorial, boat, cis/trans) using skeletal structures, Newman projections, wedge-dash, sawhorse. Identify
Lecture C2 Microscopic to Macroscopic, Part 2: Intermolecular Interactions. Let's get together.
 Lecture C2 Microscopic to Macroscopic, Part 2: Intermolecular Interactions Let's get together. Most gases are NOT ideal except at very low pressures: Z=1 for ideal gases Intermolecular interactions come
Lecture C2 Microscopic to Macroscopic, Part 2: Intermolecular Interactions Let's get together. Most gases are NOT ideal except at very low pressures: Z=1 for ideal gases Intermolecular interactions come
Pose and affinity prediction by ICM in D3R GC3. Max Totrov Molsoft
 Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
GEM4 Summer School OpenCourseWare
 GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Structure-Activity Modeling - QSAR. Uwe Koch
 Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity