Structural stability and thermodynamics of CrN magnetic phases from ab initio and experiment
|
|
|
- Meredith Golden
- 6 years ago
- Views:
Transcription
1 Structural stability and thermodynamics of CrN magnetic phases from ab initio and experiment Liangcai Zhou 1,*, David Holec 2, Matthias Bartosik 1,4, Fritz Körmann 3, Blazej Grabowski 3, Jörg Neugebauer 3 and Paul H. Mayrhofer 1,4 1 Institute of Materials Science and Technology, Vienna University of Technology, A-1040 Vienna, Austria 2 Department of Physical Metallurgy and Materials Testing, Montanuniversität Leoben, A-8700 Leoben, Austria 3 Max-Planck-Institut für Eisenforschung GmbH, D Düsseldorf, Germany 4 Christian Doppler Laboratory for Application Oriented Coating Development at the Institute of Materials Science and Technology, Vienna University of Technology, A-1040 Vienna, Austria The dynamical and thermodynamic phase stabilities of the stoichiometric compound CrN including different structural and magnetic configurations are comprehensively investigated using a first-principles density-functional-theory (DFT) plus U approach in conjunction with experimental measurements of the thermal expansion. Comparing DFT and DFT+U results with experimental data reveals that the treatment of electron correlations using methods beyond standard DFT is crucial. The non-magnetic face-centered cubic B1-CrN phase is both, elastically and dynamically unstable, even under high pressure, while CrN phases with non-zero local magnetic moments are predicted to be dynamically stable within the framework of the DFT+U scheme. Furthermore, the impact of different treatments for the exchange-correlation (xc)-functional is investigated by carrying out all computations employing the local density approximation and generalized gradient approximation. To address finite-temperature properties, both, magnetic and vibrational contributions to the free energy have been computed employing our recently developed spin-space averaging method. The calculated phase transition temperature
2 between low-temperature antiferromagnetic and high-temperature paramagnetic (PM) CrN variants is in excellent agreement with experimental values and reveals the strong impact of the choice of the xc-functional. The temperature-dependent linear thermal expansion coefficient of CrN is experimentally determined by the wafer curvature method from a reactive magnetron sputter deposited single-phase B1-CrN thin film with dense film morphology. A good agreement is found between experimental and ab initio calculated linear thermal expansion coefficients of PM B1-CrN. Other thermodynamic properties, such as the specific heat capacity, have been computed as well and compared to previous experimental data.
3 1. INTRODUCTION Transition metal nitrides have attracted much interest due to their excellent performance in applications such as hard protective coatings on cutting tools, diffusion barriers, and wear resistant electrical contacts [1-6]. Among the transition metal nitride family, chromium nitride (CrN) is valued especially for its good wear, corrosion and oxidation resistance [3, 4, 6]. There are many experimental reports on CrN due to its wide applicability in the industrial area [3, 7-17]. At room temperature, it adopts a paramagnetic (PM) cubic B1 (NaCl prototype, space group Fm3m) structure with lattice constant a=4.14 Å [12]. Upon cooling below the Néel temperature (T N = K) [7, 12, 15, 18], a simultaneous structural and magnetic phase transition to an antiferromagnetic orthorhombic (AFM Ortho, space group Pnma) phase induced by magnetic stress takes place, which is accompanied by a discontinuous volume reduction of ~0.59% [7]. The AFM Ortho-CrN phase shows a small structural distortion from the underlying cubic B1 lattice characterized by the angle α 88.3 and the AFM ordering consists of alternating double (110) planes of Cr atoms with spin up and spin down, respectively [12]. Regarding theoretical investigations, CrN has received great attention mainly due to its electronic and magnetic properties [19-26], while only a few papers have been devoted to its phase stability [22, 27]. Most theoretical studies assumed CrN in the non-magnetic rather than in the paramagnetic (PM) state [16, 28-32], while experiments clearly reveal non-vanishing local magnetic moments even above the Néel temperature [33]. The reason for this serious approximation is largely related to the challenges in describing the dynamic magnetic disorder in
4 the paramagnetic state. Recently, Alling et al. [22] employed the special quasi-random structures (SQS) approach [34] to simulate the magnetic disorder in PM B1-CrN and concluded that the magnetic disorder together with strong correlation effects in the PM B1-CrN phase largely influence the Gibbs energy. Including the magnetic entropy, a phase transition between the high-temperature PM B1-CrN phase and the low-temperature antiferromagnetic (AFM) ground state with a distorted orthorhombic (Ortho) structure was predicted at 498 K [22]. In their treatment, however, the vibrational contribution to the free energy was not taken into account, despite the known significant influence on thermodynamic properties [35]. Very recently, the same group accomplished to include vibrational contributions via disordered-local-moment molecular-dynamics-simulations in conjunction with the temperature-dependent effective potential (DLM-MD-TDEP) method [36]. The underlying DFT calculations were based on the local density approximation (LDA) with an on-site Coulomb interaction term, U. An improved phase transition temperature of 381 K has been found, which is, however, still ~100 K higher than the experimental observations. Despite the recent progress, many questions are still unanswered. A decisive issue is for instance the impact of the underlying exchange-correlation (xc)-functional on the thermodynamic stability, which is so far not known. Recently, we have developed an alternative method to compute the vibrational Gibbs energy contributions based on a spin-space averaging (SSA) method [37]. Our method does not require computationally demanding molecular dynamics simulations and can be therefore efficiently employed to scan the influence of different xc-functionals as will be performed in the present work.
5 When discussing phase stability, it is important to distinguish energetic (meta)stability, mechanical and dynamical stability. The first one is related to the Gibbs free energy, G, which predicts the phase with the lowest G to be the most stable one, all other phases being metastable or even instable. Regarding mechanical stability the Born-Huang criteria [38] for elastic constants have to be fulfilled. The dynamical stability considers the complete vibrational spectrum of a material; a material is (dynamically) stable when no imaginary phonon frequencies exist. In other words, the Born-Huang mechanical stability criteria provide a necessary condition for the dynamical stability, but not a sufficient one. For instance, by explicitly computing phonon dispersions, imaginary frequencies can occur at the Brillouin zone boundary (indicating dynamical instability), even if the Born-Huang criteria are fulfilled [39]. It is therefore necessary to perform explicit phonon calculations to evaluate the dynamical stability. Such an evaluation of the dynamical stability is computationally much more expensive as calculating the full tensor of elastic constants (in particular for systems with large numbers of atoms in their unit cells). Consequently, explicit phonon computations have been so far mostly neglected and we therefore present here a complete study of CrN phase stabilities. The nonmagnetic (NM) B1-CrN phase has been reported to be energetically unfavorable with respect to other spin arrangements with non-zero local magnetic moments [22]. It is however not clear whether NM B1-CrN is dynamically unstable with respect to mechanical distortions, i.e. whether the elastic constants fulfill criteria for mechanical stability [38] and/or if there are any imaginary frequencies in its phonon dispersion. Another question of interest is
6 whether the NM B1-CrN can be stabilized, e.g. by high pressure, and thus represents a potential metastable state. The literature data based on density functional theory (DFT) suggest its mechanical stability [28, 29, 31, 32], but an explicit evaluation via the full phonon spectrum analysis will be presented for the first time in the present paper. As mentioned above, another important and so far unresolved issue is the impact of strong electron correlation effects on the stability of CrN phases. For example, it has been argued that LDA+U is more suitable than the generalized gradient approximation (GGA) [22], but a comprehensive study employing GGA+U, which might turn out to be even more appropriate, is so far missing. Additionally, the impact of the previously proposed value for the on-site Coulomb interaction parameter U on the vibrational free energy has not been discussed so far as well [36]. The aims of the present paper can be hence summarized as follows: (1) To comprehensively investigate the energetic, mechanical and dynamical stability of CrN including non-magnetic, anti-ferromagnetic, ferromagnetic and paramagnetic configurations, (2) to elucidate the impact of the chosen xc-functional approximation and U-parameter, and (3) to compute the thermodynamic stability of the different variants. Firstly, elastic constants are calculated and used to evaluate the mechanical stability based on the Born-Huang mechanical stability criteria. As discussed above, these stability criteria might be not sufficient. Therefore, in a second step, phonon calculations are performed to prove the dynamical stability. Eventually, Gibbs free energies based on the SSA formalism [37] including vibrational and magnetic
7 contributions are computed to predict the phase transition in CrN employing GGA, GGA+U and LDA+U approaches, and the thus obtained transition temperatures are compared with available experimental results. Furthermore, thermodynamic properties such as lattice thermal expansion coefficient and specific heat capacity of the high-temperature PM B1-CrN are presented and, where applicable, compared to experimental data. In particular, we compare our DFT computed linear thermal expansion coefficient with a set of newly determined experimental data obtained by the wafer curvature method and from a single-phase face-centered cubic B1-CrN thin film. II. Methods A. Static DFT calculations As discussed above, unlike other early transition metal nitrides, CrN requires extra efforts due to the non-zero local magnetic moments of the Cr atoms. In this work the SQS approach [22, 34, 40] is used to model the PM state by employing 2x2x2 cubic supercells with 32 Cr and 32 N atoms and randomly distributing the spin-up and spin-down moments on the Cr-atoms. In order to provide a complete picture of phase stabilities, we consider in the present work also B1-CrN with ferromagnetic (FM) and AFM ordering consisting of alternating single (001) planes of Cr atoms with spin up and spin down in addition to the NM B1, PM B1 and AFM Ortho structures of CrN (cf. the crystal structures in Fig. 1). All first-principles calculations are based on DFT as implemented in the Vienna Ab initio Simulation Package (VASP) [41, 42]. The ion-electron interactions are described by the
8 projector augmented wave method (PAW) [43] with a plane wave energy cutoff of 500 ev. The semi-core p states are treated as valence for Cr (3p 6 3d 5 4s 1 ) while there are 5 valence electrons for N (2s 2 2p 3 ). In order to take into account the strong on-site Coulomb interaction (U) caused by the localized 3d electrons of Cr, the local density approximation (LDA) and the generalized gradient approximation (GGA) plus a Hubbard U-term method is adopted within the framework of the Dudarev formulation [44, 45]. Here, only the difference U-J, with J being the screened exchange energy, determines the material properties. Alling et al. [22] tested U-J in the range from 0 to 6 ev in their previous LDA+U study, and found U-J = 3 ev to yield an optimal description of the structural and electronic properties of AFM and PM-CrN. Apart from structural and electronic properties, the inclusion of the U-parameter is also decisive for the correct description of the magnetism in CrN. This is exemplified in Fig. 1, where we show that a too small value of the U-J parameter might even yield a wrong magnetic ground state for CrN. The total energy was evaluated for a number of structures as function of U-J ranging from 0 to 6 ev. For the sake of clarity, only a few structures are listed. It becomes obvious that both, standard LDA and GGA-PBE [46] predict the NM Bh-CrN phase (TaN prototype, space group C2mm) to be the ground state of CrN, which disagrees with experimental observations of AFM Ortho-CrN [7]. The energy difference between AFM Ortho-CrN and AFM B1-CrN becomes smaller as the U-J value increases. Our results suggest that the value of U-J should be larger than 1.5 ev (LDA) or 0.25 ev (GGA) in order to obtain AFM Ortho-CrN as the ground state. Following the results of Alling et al. [22], we adopted the value of U-J=3 ev also for our DFT+U calculations. In addition, the conventional GGA-PBE (without the +U term) is used in
9 the present work to demonstrate the impact of the on-site Coulomb interaction on the vibrational properties. The energy convergence criterion for electronic self-consistency was set to 0.1 mev/atom. The Monkhorst-Pack scheme [47] was used to construct k-meshes of 3x3x3 (128-atom supercells), 3x4x5 (96-atom supercells), 5x5x5 (64-atom supercells), 12x12x12 (8-atom cells), and 21x21x21 (2-atom cells). The elastic constants were calculated using the stress-strain approach discussed in detail in our previous work [40]. B. Phonon calculations The phonon calculations for NM, FM and AFM B1-CrN were performed employing 2x2x2 supercells consisting of 64 atoms constructed from a conventional face-centered cubic cell with 8 atoms. For AFM Ortho-CrN, 2x3x2 supercells with 96 atoms were created from its unit cell with 8 atoms. Super cell size convergence tests were performed for NM and FM B1-CrN using up to 4x4x4 supercells (based on primitive cells with 2 atoms) containing 128 atoms. The 64-atoms SQS adopted to simulate the paramagnetic state in B1-CrN was also used for the corresponding phonon calculations. Note that the computation of vibrational properties for paramagnetic materials is a tremendous task due to the delicate coupling of magnetic and atomic degrees of freedom [48]. We use here our recently developed statistical spin space averaging (SSA) procedure [37], which allows the computation of forces in paramagnetic materials. Within the SSA approach, the SSA force on an atom j is given as the gradient on the SSA free energy surface F SSA as [37]:
10 F SSA SSA j m j j m F HF p F {( R j), m)}, (1) R where HF j j m F {( R ), )} are the Hellmann-Feynman forces for an individual magnetic configuration m and p m B E 0 {( Rj), m)} exp[ ] denotes the Boltzmann weights. As kt B discussed in Ref. [37], the summation over different magnetic configurations, m, in the above equation can be expressed by a sum over lattice symmetry operations. In the present work we employ such a summation over lattice-symmetry equivalent forces. After applying the symmetry operations, these forces correspond to locally inequivalent magnetic configurations and allow one to perform the SSA procedure based on a single magnetic SQS structure. In the PM regime it is typically sufficient to restrict the sum in Eq. (1) to completely disordered configurations, since they dominate the partition sum (i.e. the weights p m ). Usually about magnetic configurations yield converged forces [37]. Note that the treatment of magnetic partially disordered configurations requires advanced sampling techniques, which are beyond the scope of the present manuscript and will be discussed elsewhere [49]. In the present work the magnetic disordered configurations m are constructed by all 3N Cartesian (positive and negative) displacements for the given SQS super-cell, resulting in 384 magnetic configurations. For the purpose of comparison, we additionally employed the conventional method for computing PM phonons [50], i.e. we allow atomic relaxations in the PM state which results in virtual displacements at T=0 K. The consequences of this approach and the necessity to employ the SSA averaging technique will be discussed later. Also the impact of choosing a different SQS
11 will be discussed. The finite-displacement method implemented in the phonopy [51] combined with VASP was used to calculate the real-space force constants and corresponding phonon properties. The residual forces (background forces) in the unperturbed super cell were subtracted from the force sets of the displaced structures. The summation over lattice symmetry equivalent forces according to the SSA scheme [37] has been carried out by phonopy software [52]. In order to benchmark the direct-force constant method, the NM B1-CrN, FM B1-CrN, AFM B1-CrN and AFM Ortho-CrN real-space force constants were additionally calculated within the density functional perturbation theory framework [53] as implemented in the VASP code. The phonon frequencies were subsequently calculated using the phonopy code. The phonon densities of states (DOS) from both methods (direct force constant and perturbation theory) coincide without any noticeable differences (0.5 mev/atom and 2 mev/atom difference in the free energy evaluated using both methods at 0K and 1000K, respectively). Anharmonic phonon-phonon interactions which can become important close to the melting point [54] are not relevant for the present study. We will show this explicitly in Sec. C by comparing our calculations with the molecular dynamics simulations performed in Ref. [36], which implicitly include anharmonicity. When LDA+U or GGA+U is used to treat the strong correlation effects, a small band gap opens for AFM Ortho-CrN and PM B1-CrN. Consequently, the dipole dipole interactions including properties of the Born effective charge tensor and dielectric tensor, which result in a longitudinal-transversal optical phonon branches (LO-TO) splitting [53], should be taken into account during the phonon calculations. For that purpose the dipole-dipole interactions were
12 calculated from the linear response method within the density functional perturbation theory framework as implemented in VASP at the -point of reciprocal space. The contribution of non-analytical term corrections to the dynamical matrix developed by Wang et al. was considered by the following formula [55]: jk 14 2 [ ( )] [ ( )] (, ) jk e (, ) qz j M P M P qz k, (2) N V q q where jk is the contribution from short-range interactions based on super-cell, N is the number of primitive unit cells in the super cell, V is the volume of the primitive unit cell, q is the wave vector, and are the Cartesian axes, Z*(j) is the Born effective charge tensor of the j th atom in the primitive unit cell, and is the high frequency static dielectric tensor, i.e., the contribution to the dielectric permittivity tensor from the electronic polarization. C. Thermodynamic properties Once the phonon DOS is obtained, the vibrational energy and its effect on thermal properties can be directly evaluated. In combination with first-principles calculations, the Helmholtz free energy, F( VT, ), is the most convenient choice for a thermodynamic potential, since it is a natural function of V and T: F(V,T ) E 0 (V ) F el (V,T ) F vib (V,T ) F mag (V,T ), (3) where E0( V) is the internal energy at 0 K obtained from the equation of state [56], F el (V,T ) and F vib (V,T ) are the thermal electronic and lattice contributions to the free energy, respectively. Further details on the ab initio calculations of F el (V,T ) and F vib (V,T ) are
13 discussed in Ref. [57] and references therein. F mag (V,T) is the magnetic free energy which is in the present work considered within the mean-field approximation as [58]: F mag k B T ln(m (T,V ) 1), (4) where M(T,V) is the magnitude of the local magnetic moment (in units of B ) and k B is the Boltzmann constant. In this work we used the averaged magnitude of the local magnetic moment, M(T=0 K,V=V 0 ), at 0 K at the ground state volume, V 0, to predict the magnetic entropy. Our test calculations revealed that the inclusion of the volume dependence of M(T,V) has no significant impact on our main results and conclusions, and will be therefore not considered in the following. The average magnetic moments based on GGA, GGA+U, and LDA+U schemes, which are in the following used in Eq. (4), are 2.48 B, 2.94 B, and 2.83 B, respectively, which agrees well with previous theoretical values [22, 27]. D. Experimental details Since no experimental study has been devoted so far to the lattice thermal expansion coefficient,, this property has been measured within the present work. A single-phase face-centered cubic CrN thin film was deposited on Si (100) substrates (7 x 21 mm 2 ) using reactive magnetron sputtering. The deposition was carried out at a deposition temperature of 743 K in an Ar and N 2 gas atmosphere of a total pressure of 0.4 Pa and a constant Ar/N 2 flow ratio of 2/3. A target power of 250 W and a 3 Cr target (purity 99.9 %) were used. During the deposition, a bias potential of 70 V was applied to the substrates to ensure dense film morphology. The film thickness was measured using cross-sectional scanning electron
14 microscopy. The biaxial stress in CrN was recorded as a function of temperature using the wafercurvature method [59]. The wafer-curvature system was operating with an array of parallel laser beams and a position sensitive charge-coupled device detector. The sample was heated by a ceramic heating plate with a constant heating rate of 5 K/min from room temperature to 518 K under vacuum conditions of 10-4 mbar. The maximum temperature was chosen to be clearly below the deposition temperature to avoid thermally activated processes in the film (e.g. recovery of deposition-induced defects) and the substrate and thus to guarantee pure thermoelastic behavior. The film stress was deduced from the sample curvature 1/R according to: 2 Mh, (5) 6Rt f with M ~ 180 GPa being the biaxial modulus of the substrate, h = 380 m the substrate and t f = 1.50 m the film thicknesses. Due to the mismatch in the coefficients of thermal expansion of CrN and Si, the temperature change results in the formation of thermal stresses in the film. When the elastic moduli are taken as constants in the given temperature range, the linear thermal expansion coefficient,, of CrN thin film is related to that of substrate (Si) as: ( T) ( ) 1 v d CrN si(001) T, (6) ECrN dt with E CrN =330 GPa, v CrN =0.22 [60, 61] and the thermal expansion coefficient of substrate (Si) is expressed as [62]: 3 T T 4 T si(001) (3.725[1 exp( ) ]) 10 6 T in K. (7)
15 III Results and Discussion A. Elastic constants and mechanical stability The elastic constants of the different studied structures as calculated by the GGA, GGA+U, and LDA+U schemes are listed in Table I. It can be seen that most of the elastic constants derived from GGA and GGA+U are smaller than those obtained from the LDA+U scheme. This is consistent with the fact that GGA frequently overestimates lattice parameters leading to an underestimation of binding energies, elastic properties, and phonon frequencies for most materials [63]. In contrast, LDA (or LDA+U) tends to overestimate binding as expressed by smaller lattice parameters, and higher elastic constants and phonon frequencies when compared with experimental or GGA and GGA+U values. A comparison with the limited available experimental data reveals good agreement for the C 11 component (in particular for GGA+U), while C 12 and C 44 seem to be overestimated by all the LDA+U, GGA and GGA+U theoretical predictions. However, it might be that the substantially too low experimental C ij constants are a consequence of the fact that the measurements have been performed on a polycrystalline sample at room temperature, i.e., the measured elastic constants may be influenced by soft grain boundaries [61]. The mechanical stability of any crystal requires the strain energy to be positive, which implies that the whole set of elastic constants, C ij, must satisfy the Born-Huang stability criterion
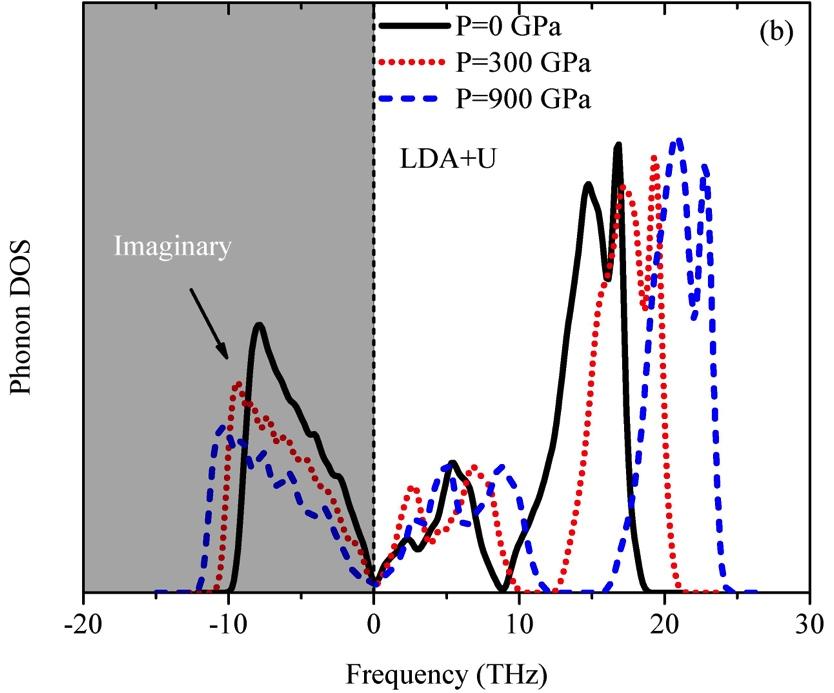
16 [38]. Using this requirement Table I shows that CrN is mechanically stable within the GGA scheme for all magnetic and structural configurations studied here in consistency with previous results [28, 29, 32]. In contrast, we observe that C 44 derived from the LDA+U and GGA+U schemes suggest NM B1-CrN to be mechanically unstable (C 44 < 0). This result underlines the importance of strong correlation effects, which were not considered in previous elastic constant calculations using standard LDA or GGA approaches. Finally, we observe AFM B1-CrN to show a small tetragonal distortion, which is reflected in the number of nonequivalent elastic constants: C 11, C 12, C 13, C 33, C 44 and C 66 in agreement with previous results [28]. B. Dynamical stability As discussed above, a necessary condition for a structure to be dynamically stable is that it is stable against all possible small perturbations of its atomic structure, i.e., that all phonon frequencies must be real. The GGA, GGA+U, and LDA+U phonon spectra of NM B1-CrN are presented in Fig. 2 (a). Imaginary acoustic branches at the X and W-points of the Brillouin zone suggest an internal instability of NM B1-CrN. These phonon anomalies are due to the high electron DOS at the Fermi level of NM B1-CrN [19], which causes the existence of soft phonon modes at these points. We evaluated the phonon density of states (DOS) of NM B1-CrN for several pressures up to 900 GPa to check whether applying external pressure can eliminate these imaginary phonons. For the sake of clarity, only the representative LDA+U results are presented in Fig. 2 (b), yielding the following tendency: NM B1-CrN is dynamically unstable even under high pressures, indicating the NM B1-CrN cannot be stabilized by high pressure.
17 In order to check the phase stability of CrN for various magnetic states, the phonon spectra of CrN from GGA, GGA+U, and LDA+U with ordered magnetic states are presented in Fig. 3. The comparison of the phonon spectra of FM B1-CrN, AFM B1-CrN and AFM Ortho-CrN from GGA, and GGA+U or LDA+U reveals that the phonon spectra are significantly shifted to higher frequencies for the latter approximations. The GGA and GGA+U based phonon frequencies are always lower than those derived from the LDA+U scheme, a trend already reflected by the softer elastic constants when GGA or GGA+U is used. For FM and AFM B1-CrN, when no U-correction is used, a softening tendency for the phonon branch around the X-point is observed, and for AFM B1-CrN phonon frequencies even decrease to negative values. The dynamical instability, not observed via the elastic constants calculations, indicates once more the necessity of explicit phonon calculations for the stability analysis. This phonon anomaly observed for the GGA calculations originates from the high electron DOS at the Fermi level. When the on-site Coulomb repulsion U in the DFT+U scheme is switched on, the electron density of states at Fermi level significantly decreases for the FM B1-CrN although no gap opens [21]. Here, one should note that despite the fact that the DFT+U approximation does not result in a gap opening at the Fermi level for NM- and FM B1-CrN, as shown in Refs. [19, 21], it significantly increases the stability of FM and AFM B1-CrN as measured by phonon frequencies around X-point. No imaginary frequencies in the phonon dispersion curves are observed independent of whether the GGA+U or LDA+U scheme is used, implying that FM B1-CrN, AFM B1-CrN and AFM Ortho-CrN are all dynamically stable, and represent hence potential (meta)stable phases.
18 In contrast to the ordered magnetic states of FM B1-CrN, AFM B1-CrN and AFM Ortho-CrN, the magnetic state in PM B1-CrN is disordered. As a first step we allow for atomic relaxations at 0 K. Since at finite temperatures the magnetic fluctuations are usually faster compared to the atomic motion, these displacements can be considered as artificial. In order to test the effect of such artificial static displacements, the atomic positions are fixed to the ideal B1 sites and cell shape remains cubic (we refer to this structure as unrelaxed hereafter). The phonon DOS corresponding to unrelaxed and fully relaxed SQS are presented in Fig. 4 (a). Clearly, the phonon DOS of the unrelaxed SQS exhibits some different features than that of the fully relaxed one, which means that the artificial static displacements have some impact on the phonon calculations. A closer inspection of the phonon dispersion of the unrelaxed structure reveals some imaginary phonon modes at the point. In order to elucidate if these imaginary frequencies are physical, we employ in the following the recently developed SSA method, Eq. (1), to compute the phonon frequencies in the paramagnetic regime. In fact, no imaginary phonon frequencies are obtained (see the red dashed line in Fig. 5) revealing the applicability of the method and the fact that the imaginary phonon modes along the acoustic dispersions are indeed caused by the artificial T=0 K relaxations. In order to further analyze the impact of artificial T=0 K relaxations on the specific SQS structure, we have performed similar calculations for a second SQS with a different spin arrangement. Figure 4 (b) shows that the phonon DOS s for the two SQS almost fully coincide, suggesting that a particular spin arrangement and consequently a specific spin-induced relaxation of atoms at T=0 K in the SQS of PM B1-CrN does not significantly affect the phonon DOS and hence thermodynamic
19 properties. Since the SSA computed phonon DOS does not show any imaginary frequencies and does not require any (unphysical) T=0 K relaxations, we will proceed with the SSA obtained phonon DOS and thermodynamic properties for the following thermodynamic analysis. As discussed above, super cell convergence tests for NM and AFM variants have revealed that a 2x2x2 super-cell is sufficient for an accurate description within the considered temperature range. This is in agreement with the findings of Shulumba et al. [36], who reported convergence with respect to the super cell size for PM B1-CrN for a super cell with 64 atoms (2x2x2), which we will employ in the following. Figure 5 presents the phonon spectra of PM B1-CrN derived from GGA, GGA+U and LDA+U schemes employing the SSA method, and in comparison with available experimental data (open circles [23] ) at the G point. It demonstrates that the transverse branches derived from DFT+U are significantly shifted to higher values and the LO-TO splitting happens at the G point, when comparing to the result from GGA scheme. The obtained dispersion from DFT+U is in excellent agreement with experimental data (blue circles), especially the results derived from the GGA+U scheme are very close to experimental data. The accurately predicted transverse and longitudinal optical phonon frequencies at the G point further validate the reliability of the SSA scheme. The phonon spectra derived from GGA, GGA+U, and LDA+U in combination with the SSA scheme confirm that PM B1-CrN is dynamically stable. C. Thermodynamic stability and thermodynamic properties of CrN
20 The Gibbs free energy has been evaluated according to the treatment described in Sec. C employing in particular the SSA method for the PM phases. Due to the opening of a small gap in AFM Ortho-CrN and PM B1-CrN as obtained from GGA+U and LDA+U [24], the thermal electronic contribution F el (V,T ) to the Helmholtz free energy can be ignored for AFM Ortho-CrN and PM B1-CrN. Firstly, we compare the SSA results for the Gibbs energies to previous results from the DLM-MD-TDEP method [36] to verify the reliability of our method and also to check the influence of anharmonic effects on the vibrational free energy. The comparison between the present work and the results from DLM-MD-TDEP method [36] is presented in Fig. 6. As the latter method is based on MD simulations, anharmonic contributions are implicitly included. This allows us to evaluate the importance of anharmonic effects for the considered temperature regime. Figure 6 demonstrates that the vibrational free energies of PM B1-CrN from both methods (SSA and DLM-MD-TDEP) fully coincide with each other and that there is no noticeable difference even at high temperatures. The largest deviation of about 5 mev/atom is observed for AFM Ortho-CrN at 1000 K. Keeping in mind that the phase transition under consideration occurs at room temperature, we obtain an excellent agreement with the previous work in the relevant temperature regime revealing that anharmonic contributions are not decisive for the considered transition. Next we concentrate on the impact of the xc-functional. The differences of harmonic vibrational free energies as a function of temperature between AFM Ortho-CrN and PM B1-CrN
21 in the GGA, GGA+U and LDA+U schemes are presented in Fig. 7 (a). For all considered scenarios, the vibrational contribution to the free energy favors the PM B1-CrN phase. This contribution becomes smaller when the strong correlation effects are taken into account by using Hubbard Coulomb term U. We now include the magnetic entropy, Eq. (4), into our free energy computations. The results including both, vibrational as well as the magnetic entropy terms evaluated using GGA, GGA+U and LDA+U schemes, are presented in Fig. 7 (b) as a function of temperature for AFM Ortho-CrN and PM B1-CrN phases. The critical temperature for the structural transition between AFM Ortho and PM B1 CrN is Ts=428 K for GGA, Ts=300 K for GGA+U, and Ts=370 K for the LDA+U scheme. The excellent agreement of the latter with the previous theoretical results [36] reveals once more the reliability of our SSA approach. A particularly important conclusion is that the GGA+U value turns out to be significantly closer to the experimental value compared to the previous LDA+U estimations. This improvement of GGA+U compared to the LDA+U data is remarkable and of similar magnitude and importance as the inclusion of vibrational contributions [22,36]. The inclusion of vibrational free energy contributions is considerable and shifts the transition temperature, e.g. for GGA, by almost 500 K [22]. The inclusion of magnetic entropy turns out to be similarly important. To elucidate the impact of magnetic contributions, we included in Fig. 7 (b) also the Gibbs free energy curves without the magnetic entropy term S mag in Eq. (4) (dotted lines). Consequently, AFM Ortho-CrN becomes energetically preferred over
22 the PM B1-CrN phase in the whole investigated temperature range. In agreement with previous works [22, 36] we can therefore conclude that both, magnetic and vibrational contributions are equally important to accurately predict the transition temperature. The remaining difference between predicted and experimental values for the phase transition temperature may be related to the approximate treatment of magnetism (neglected non-collinear magnetic configurations, magnetic entropy extracted from mean-field approximation) or to the inherent DFT approximation, i.e. the treatment of the xc-functional. The phase transition from AFM Ortho-CrN to PM B1-CrN induced by temperature can be alternatively triggered by pressure. In order to investigate the pressure effect on the phase transition, the Gibbs energy is computed via the Legendre transformation as GpT (, ) FVp ( ( ), T) pvpt (, ) (8) where F is the Helmholtz free energy, Eq. (3), evaluated within the framework of the quasi-harmonic approach at several volumes including the magnetic entropy contributions from Eq. (4) for PM B1-CrN. p is the pressure and V( pt, ) is the volume as a function of pressure and temperature. Using the Gibbs energies of AFM Ortho-CrN and PM B1-CrN based on the GGA+U and LDA+U schemes, the p-t phase diagram was derived (Fig. 8). The transition temperature increases with pressure in agreement with experimental and theoretical results reported in literature. The transition temperature at p=0 GPa obtained from experiment [7, 12, 15] varies from 200 K to 287 K, showing an uncertainty of up to 87 K. Within this error-bar, our theoretical predictions based on the GGA+U method are in excellent agreement with the
23 experimental work and yielding a significant improvement as compared with the previously reported theoretical results. It should be noted that the transition temperatures at p=0 GPa are approximately 10 K lower than the results in Fig. 7. This is due to the fact that the free energy in Eq. (3) is evaluated within the quasi-harmonic approximation, while the results presented in Fig. 7 are obtained from the harmonic approach. This implies that the quasi-harmonic contribution (i.e., caused by thermal expansion) is not as important as for example the strong correlation effect or the magnetic entropy contribution, within the considered temperature-pressure regime. On the other hand, thermal expansion is a key thermodynamic property of PM B1-CrN when it is applied as a high temperature coating material. If compared to experiment, the derivative of the linear thermal expansion coefficient, (T), provides an even more sensitive measurement of the accuracy achievable by the theoretical predictions. The calculated (T) of PM B1-CrN is compared to our experimental data in Fig. 9. The results are in good agreement around room temperature, and the discrepancy increases with raising temperature. It was recently demonstrated that (T) of CrN strongly depends on the grains size [64]. Depending on the thin film microstructure, (T) varies in the range of /K and /K, which for this material somewhat complicates a distinct comparison with our predictions. Considering these complications, the overall agreement with experiment is very reasonable. Finally, in Fig. 10 we plot the specific heat capacity, C P (T), at constant (ambient) pressure as a function of temperature predicted from our GGA+U and LDA+U calculations, together with available experimental data from literature [65]. The GGA+U values are slightly closer to the
24 experimental data. This is consistent with the findings for the phase transition temperature discussed above; however, the differences between LDA+U and GGA+U are only minor. The literature data exhibit a peak near the phase transition temperature (~280 K), due to the Néel transition, which has not been accounted for in the present study. This peak originates from the magnetic phase transition and can be resolved only when the magnetic contribution is exactly evaluated by, for example the Heisenberg model for spin interactions [66]. Nevertheless, our predictions are reasonably accurate from 0K to about 200K and also above 600K. IV. Conclusions The phase stability of different structural and magnetic configurations of stoichiometric CrN is studied systematically by first-principles calculations based on the GGA, GGA+U, and LDA+U schemes. In combination with our recently developed SSA procedure, the phonon contributions in paramagnetic materials are computed. A comparison of the three exchange-correlation approximations demonstrates, that strong correlation effects have a significant impact on the mechanical and phase stability of CrN. The elastic constants and phonon spectra show that the non-magnetic B1-CrN phase is dynamically unstable even under high pressures, due to the high electron density of states at Fermi level. The (meta)stability of the ferromagnetic and antiferromagnetic B1-CrN phase is significantly improved when strong correlation effects are considered using the DFT+U approach. Including the vibrational, electronic and magnetic free energy contributions, the results of our LDA+U-SSA based approach agree well with previous LDA+U-MD simulations. By performing
25 for the first time finite-temperature GGA+U simulations for CrN we show that the treatment of the xc-functional, i.e. GGA+U vs. LDA+U, is decisive to predict accurate phase transition temperatures in CrN. In particular we find that GGA+U significantly improves previous LDA+U predictions. The phase transition between AFM Ortho-CrN and the paramagnetic B1-CrN phase is predicted to be 293 K at ambient pressure being in excellent agreement with the experimental value of K. The impact of the xc-functional is similar in magnitude to the impact of vibrational contributions to the phase stability. The linear thermal expansion coefficient, (T), and the heat capacity, C p, of PM B1-CrN as a function of temperature is obtained from experimental measurements and ab initio calculations. The comparison between the experimental results and predictions for these thermodynamic properties reveals good agreement and further confirms the reliability of our theoretical method. ACKNOWLEDGEMENTS The financial support by the START Program (Y371) of the Austrian Science Fund (FWF) and the Austrian Federal Ministry of Economy, Family and Youth and the National Foundation for Research, Technology and Development is gratefully acknowledged. Funding by the European Research Council under the EU`s 7th Framework Programme (FP7/ )/ERC Grant agreement and by the collaborative research center SFB 761 Stahl - ab initio of the Deutsche Forschungsgemeinschaft is also gratefully acknowledged. First-principles calculations were carried out partially on the cluster supported by the Computational Materials Design department at the Max-Planck-Institut für Eisenforschung GmbH in Düsseldorf and the Vienna Scientific Cluster (VSC). The authors are thankful to Dr. Yuji Ikeda from Kyoto University for valuable help with the phonopy software.
26 References [1] L. Hultman, Vac. 57, 1 (2000). [2] W.-D. Munz, J. Vac. Sci. Technol. A 4, 2717 (1986). [3] B. Navinšek, and P. Panjan, Surf. Coat. Technol. 97, 182 (1997). [4] P. H. Mayrhofer, H. Willmann, and C. Mitterer, Surf. Coat. Technol , 222 (2001). [5] W. R. L. Lambrecht, M. S. Miao, and P. Lukashev, J. Appl. Phys. 97, 10D306 (2005). [6] G. Berg et al., Surf. Coat. Technol. 86, 184 (1996). [7] L. M. Corliss, N. Elliott, and J. M. Hastings, Phys. Rev. 117, 929 (1960). [8] A. Ney et al., Appl. Phys. Lett. 89, (2006). [9] K. Inumaru et al., Phys. Rev. B 75, (2007). [10] P. H. Mayrhofer et al., Scr. Mater. 57, 249 (2007). [11] C. X. Quintela, F. Rivadulla, and J. Rivas, Appl. Phys. Lett. 94, (2009). [12] F. Rivadulla et al., Nat. Mater. 8, 947 (2009). [13] X. Y. Zhang et al., Phys. Rev. B 83, (2011). [14] M. Chen et al., Chem. Eur. J. 18, (2012). [15] S. Wang et al., Phys. Rev. B 86, (2012). [16] Z. Zhang et al., Phys. Rev. B 87, (2013). [17] D. Gall et al., J. Appl. Phys. 91, 5882 (2002). [18] X. Y. Zhang et al., Phys. Rev. B 84, (2011). [19] A. Filippetti, W. E. Pickett, and B. M. Klein, Phys. Rev. B 59, 7043 (1999). [20] A. Filippetti, and N. A. Hill, Phys. Rev. Lett. 85, 5166 (2000). [21] A. Herwadkar, and W. R. L. Lambrecht, Phys. Rev. B 79, (2009). [22] B. Alling, T. Marten, and I. A. Abrikosov, Phys. Rev. B 82, (2010). [23] X. Y. Zhang, and D. Gall, Phys. Rev. B 82, (2010). [24] A. S. Botana et al., Phys. Rev. B 85, (2012). [25] A. S. Botana et al., Phys. Rev. B 87, (2013). [26] A. Lindmaa et al., Phys. Rev. B 88, (2013). [27] B. Alling, T. Marten, and I. A. Abrikosov, Nat. Mater. 9, 283 (2010). [28] V. Antonov, and I. Iordanova, in AIP Conference Proceedings2010), pp [29] Y. Liang, X. Yuan, and W. Zhang, Solid. State. Commun. 150, 2045 (2010). [30] H. Lin, and Z. Zeng, Chin. Phy. B 20, (2011). [31] M. G. Brik, and C. G. Ma, Comput. Mater. Sci. 51, 380 (2012). [32] Z. Y. Jiao et al., Mod. Phys. Lett. B 27, (2013). [33] C. G. Shull, W. A. Strauser, and E. O. Wollan, Phys. Rev. 83, 333 (1951). [34] S. H. Wei et al., Phys. Rev. B 42, 9622 (1990). [35] A. Van de Walle, and G. Ceder, Rev. Mod. Phys. 74, 11 (2002). [36] N. Shulumba et al., Phys. Rev. B 89, (2014). [37] F. Körmann et al., Phys. Rev. B 85, (2012).
27 [38] M. Born, K. Huang, and G. Britain, Dynamical theory of crystal lattices (Clarendon Press Oxford, 1954). [39] D. M. Clatterbuck et al., Phys. Rev. Lett. 91, (2003). [40] L. Zhou, D. Holec, and P. H. Mayrhofer, J. Appl. Phys. 113, (2013). [41] G. Kresse, and J. Hafner, Phys. Rev. B 47, 558 (1993). [42] G. Kresse, and J. Furthmüller, Phys. Rev. B 54, (1996). [43] G. Kresse, and D. Joubert, Phys. Rev. B 59, 1758 (1999). [44] V. I. Anisimov, J. Zaanen, and O. K. Andersen, Phys. Rev. B 44, 943 (1991). [45] S. L. Dudarev et al., Phys. Rev. B 57, 1505 (1998). [46] J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996). [47] H. J. Monkhorst, and J. D. Pack, Phys. Rev. B 13, 5188 (1976). [48] F. Körmann et al., J. Phys.: Condens. Matter 25, (2013). [49] G. Körmann, Dutta, Hickel, Mauger, Fultz, Neugebauer, submitted (2014). [50] K. Parlinski, Z. Q. Li, and Y. Kawazoe, Phys. Rev. Lett. 78, 4063 (1997). [51] A. Togo, F. Oba, and I. Tanaka, Phys. Rev. B 78, (2008). [52] S. Ikeda, Togo, Tanaka, submitted. (2014). [53] S. Baroni et al., Reviews of Modern Physics 73, 515 (2001). [54] B. Grabowski et al., Phys. Rev. B 79, (2009). [55] Y. Wang et al., Journal of Physics: Condensed Matter 22, (2010). [56] F. Birch, Phys. Rev. 71, 809 (1947). [57] B. Grabowski et al., Phys. Rev. B 84, (2011). [58] F. Körmann et al., physica status solidi (b) 251, 53 (2013). [59] G. C. A. M. Janssen et al., Thin Solid Films 517, 1858 (2009). [60] J. Almer et al., J. Appl. Phys. 94, 697 (2003). [61] K. J. Martinschitz et al., J. Appl. Crystallogr. 42, 416 (2009). [62] M. O, Semiconductors: Data Handbook (2004). [63] B. Grabowski, T. Hickel, and J. Neugebauer, Phys. Rev. B 76, (2007). [64] R. Daniel et al., Acta. Mater. 59, 6631 (2011). [65] M. W. Chase et al., NIST-JANAF Themochemical Tables, Fourth Edition, J. Phys. Chem. Ref. Data, (1998). [66] T. Hickel et al., J. Phys.: Condens. Matter 24, (2012).
28 Table. T=0 K elastic constants in GPa of CrN within the framework of GGA, GGA+U, and LDA+U methods including a comparison with experimental values. Structure C11 C22 C33 C12 C13 C23 C44 C55 C66 GGA NM B1 FM B1 AFM B1 PM B1 AFM Ortho GGA+U LDA+U GGA GGA+U LDA+U GGA GGA+U LDA+U GGA GGA+U LDA+U Exp. [60, 61] GGA GGA+U LDA+U
29 Figure captions FIG. 1. (Color online) T=0 K total energy of the stoichiometric compound CrN in different structural and magnetic configurations as a function of the U-J term in the (a) LDA+U and (b) GGA+U schemes. The energy of the AFM Ortho-CrN phase is used as reference. The vertical dash-dotted line indicates the U-J used in the present work. The crystal structures to the right show the various investigated atomic and magnetic arrangements with the blue (white) balls indicating Cr (N) atoms. FIG. 2. (Color online) (a) Phonon spectra of NM B1-CrN calculated using GGA, GGA+U, and LDA+U, and (b) phonon DOS as a function of pressure for NM B1-CrN from the LDA+U scheme. The gray shaded region highlights imaginary frequencies. FIG. 3. (Color online) Phonon spectra of (a) FM B1-CrN, (b) AFM B1-CrN and (c) AFM Ortho-CrN calculated using the GGA, GGA+U and LDA+U schemes. FIG. 4. (Color online) Phonon DOS s from the LDA+U scheme (a) for fully relaxed and unrelaxed SQS, (b) for two SQSs with different SRO parameters. FIG. 5. (Color online) Phonon spectra of PM B1-CrN simulated with a 64-atom SQS from the GGA, GGA+U and LDA+U schemes in combination with SSA approach, together with experiments at the G point from Raman and infrared measurements (open circles [23] ). FIG. 6. (Color online) Comparison of the vibrational free energies for AFM Ortho-CrN (blue lines) and PM B1-CrN (black lines) under the framework of LDA+U. The solid lines denote the
30 results from SSA approach in the present work and dotted lines denote DLM-MD-TDEP method [36]. FIG. 7. (Color online) (a) The differences of vibrational free energy between AFM Ortho-CrN and PM B1-CrN in the GGA, GGA+U and LDA+U schemes, and (b) calculated phase transition temperature from Helmholtz free energies of AFM Ortho-CrN and PM B1-CrN by taking lattice vibrational and magnetic contributions into account under the framework of GGA and LDA+U scheme. The dotted lines denote the Helmholtz free energies of PM B1-CrN without magnetic contributions. FIG. 8. (Color online) Calculated pressure-temperature phase diagram of CrN based on the GGA+U (black line) and LDA+U (blue lines) schemes compared to experiment (open circles [12] and squares [15]). The solid lines represent the phase diagram based on the relevant spectrum of excitations: total energy, vibrational and magnetic entropy contributions. For LDA+U, the dashed line shows the phase diagram if only the total energy and magnetic excitations are included to emphasize the impact of vibrational entropy. FIG. 9. (Color online) Linear thermal expansion coefficient, (T), of PM B1-CrN computed within the GGA+U (black solid line) and LDA+U (blue dashed line) schemes compared to experimental data (open squares) as obtained in the present work. FIG. 10. (Color online) Heat capacities of PM B1-CrN in the GGA+U (black solid line) and LDA+U (blue dashed line) schemes compared to experimental values (open triangles [65]).
31
32 (a) LDA+U (b) GGA+U Energy (ev/atom) NM B1 NM Bh FM B1 AFM B1 AFM Ortho Used U J Used U J magnetism NM B1 NM Bh FM B AFM B U J (ev) U J (ev) AFM Ortho FIG. 1

33 FIG. 2



34 FIG. 3
SnO 2 Physical and Chemical Properties due to the Impurity Doping
 , March 13-15, 2013, Hong Kong SnO 2 Physical and Chemical Properties due to the Impurity Doping Richard Rivera, Freddy Marcillo, Washington Chamba, Patricio Puchaicela, Arvids Stashans Abstract First-principles
, March 13-15, 2013, Hong Kong SnO 2 Physical and Chemical Properties due to the Impurity Doping Richard Rivera, Freddy Marcillo, Washington Chamba, Patricio Puchaicela, Arvids Stashans Abstract First-principles
Phonon calculations with SCAN
 Workshop on the SCAN density functional: Fundamentals, practices, and extensions Temple university, Philadelphia May 18th, 2017 Hands-on tutorial 3 Phonon calculations with SCAN Yubo Zhang and Jianwei
Workshop on the SCAN density functional: Fundamentals, practices, and extensions Temple university, Philadelphia May 18th, 2017 Hands-on tutorial 3 Phonon calculations with SCAN Yubo Zhang and Jianwei
Defects in TiO 2 Crystals
 , March 13-15, 2013, Hong Kong Defects in TiO 2 Crystals Richard Rivera, Arvids Stashans 1 Abstract-TiO 2 crystals, anatase and rutile, have been studied using Density Functional Theory (DFT) and the Generalized
, March 13-15, 2013, Hong Kong Defects in TiO 2 Crystals Richard Rivera, Arvids Stashans 1 Abstract-TiO 2 crystals, anatase and rutile, have been studied using Density Functional Theory (DFT) and the Generalized
CHAPTER: 8. ELECTRONIC STRUCTURE AND ELASTIC PROPERTIES OF CrC AND CrN. 8.1 Introduction. Ph.D. Thesis: J. Maibam
 CHAPTER -8 CHAPTER: 8 ELECTRONIC STRUCTURE AND ELASTIC PROPERTIES OF CrC AND CrN 8.1 Introduction In this chapter, we have selected CrC and CrN from group VIB transition metal carbides and nitrides for
CHAPTER -8 CHAPTER: 8 ELECTRONIC STRUCTURE AND ELASTIC PROPERTIES OF CrC AND CrN 8.1 Introduction In this chapter, we have selected CrC and CrN from group VIB transition metal carbides and nitrides for
Supporting information. Realizing Two-Dimensional Magnetic Semiconductors with. Enhanced Curie Temperature by Antiaromatic Ring Based
 Supporting information Realizing Two-Dimensional Magnetic Semiconductors with Enhanced Curie Temperature by Antiaromatic Ring Based Organometallic Frameworks Xingxing Li and Jinlong Yang* Department of
Supporting information Realizing Two-Dimensional Magnetic Semiconductors with Enhanced Curie Temperature by Antiaromatic Ring Based Organometallic Frameworks Xingxing Li and Jinlong Yang* Department of
Quantum Chemical Study of Defective Chromium Oxide
 , March 13-15, 2013, Hong Kong Quantum Chemical Study of Defective Chromium Oxide Richard Rivera, Soraya Jácome, Frank Maldonado, Arvids Stashans 1 Abstract Through the use of first-principles calculations
, March 13-15, 2013, Hong Kong Quantum Chemical Study of Defective Chromium Oxide Richard Rivera, Soraya Jácome, Frank Maldonado, Arvids Stashans 1 Abstract Through the use of first-principles calculations
arxiv: v1 [cond-mat.mtrl-sci] 19 Mar 2014
![arxiv: v1 [cond-mat.mtrl-sci] 19 Mar 2014](/thumbs/71/66018354.jpg "arxiv: v1 [cond-mat.mtrl-sci] 19 Mar 2014") Vibrational free energy and phase stability of paramagnetic and antiferromagnetic CrN from ab-initio molecular dynamics arxiv:1403.4766v1 [cond-mat.mtrl-sci] 19 Mar 2014 Nina Shulumba, 1 Björn Alling,
Vibrational free energy and phase stability of paramagnetic and antiferromagnetic CrN from ab-initio molecular dynamics arxiv:1403.4766v1 [cond-mat.mtrl-sci] 19 Mar 2014 Nina Shulumba, 1 Björn Alling,
Curvature-enhanced Spin-orbit Coupling and Spinterface Effect in Fullerene-based Spin Valves
 Supplementary Information Curvature-enhanced Spin-orbit Coupling and Spinterface Effect in Fullerene-based Spin Valves Shiheng Liang 1, Rugang Geng 1, Baishun Yang 2, Wenbo Zhao 3, Ram Chandra Subedi 1,
Supplementary Information Curvature-enhanced Spin-orbit Coupling and Spinterface Effect in Fullerene-based Spin Valves Shiheng Liang 1, Rugang Geng 1, Baishun Yang 2, Wenbo Zhao 3, Ram Chandra Subedi 1,
Facet engineered Ag 3 PO 4 for efficient water photooxidation
 Supporting Information Facet engineered Ag 3 PO 4 for efficient water photooxidation David James Martin, Naoto Umezawa, Xiaowei Chen, Jinhua Ye and Junwang Tang* This file includes the following experimental/theoretical
Supporting Information Facet engineered Ag 3 PO 4 for efficient water photooxidation David James Martin, Naoto Umezawa, Xiaowei Chen, Jinhua Ye and Junwang Tang* This file includes the following experimental/theoretical
SMARTMET project: Towards breaking the inverse ductility-strength relation
 SMARTMET project: Towards breaking the inverse ductility-strength relation B. Grabowski, C. Tasan SMARTMET ERC advanced grant 3.8 Mio Euro for 5 years (Raabe/Neugebauer) Adaptive Structural Materials group
SMARTMET project: Towards breaking the inverse ductility-strength relation B. Grabowski, C. Tasan SMARTMET ERC advanced grant 3.8 Mio Euro for 5 years (Raabe/Neugebauer) Adaptive Structural Materials group
Supplementary Information
 Supplementary Information Supplementary Figure 1: Electronic Kohn-Sham potential profile of a charged monolayer MoTe 2 calculated using PBE-DFT. Plotted is the averaged electronic Kohn- Sham potential
Supplementary Information Supplementary Figure 1: Electronic Kohn-Sham potential profile of a charged monolayer MoTe 2 calculated using PBE-DFT. Plotted is the averaged electronic Kohn- Sham potential
SUPPLEMENTARY FIGURES
 1 SUPPLEMENTARY FIGURES Supplementary Figure 1: Initial stage showing monolayer MoS 2 islands formation on Au (111) surface. a, Scanning tunneling microscopy (STM) image of molybdenum (Mo) clusters deposited
1 SUPPLEMENTARY FIGURES Supplementary Figure 1: Initial stage showing monolayer MoS 2 islands formation on Au (111) surface. a, Scanning tunneling microscopy (STM) image of molybdenum (Mo) clusters deposited
Supporting Information
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2015 Supporting Information Single Layer Lead Iodide: Computational Exploration of Structural, Electronic
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2015 Supporting Information Single Layer Lead Iodide: Computational Exploration of Structural, Electronic
Selectivity in the initial C-H bond cleavage of n-butane on PdO(101)
") Supporting Information for Selectivity in the initial C-H bond cleavage of n-butane on PdO(101) Can Hakanoglu (a), Feng Zhang (a), Abbin Antony (a), Aravind Asthagiri (b) and Jason F. Weaver (a) * (a)
Supporting Information for Selectivity in the initial C-H bond cleavage of n-butane on PdO(101) Can Hakanoglu (a), Feng Zhang (a), Abbin Antony (a), Aravind Asthagiri (b) and Jason F. Weaver (a) * (a)
Structural and Electronic Effects on the Properties of Fe 2 (dobdc) upon Oxidation with N 2 O
 upon Oxidation with N 2 O") Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-1 Structural and Electronic Effects on the Properties of Fe (dobdc) upon Oxidation with N O oshua Borycz, 1, oachim Paier,
Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-1 Structural and Electronic Effects on the Properties of Fe (dobdc) upon Oxidation with N O oshua Borycz, 1, oachim Paier,
Supporting Information Tuning Local Electronic Structure of Single Layer MoS2 through Defect Engineering
 Supporting Information Tuning Local Electronic Structure of Single Layer MoS2 through Defect Engineering Yan Chen, 1,2,,$, * Shengxi Huang, 3,6, Xiang Ji, 2 Kiran Adepalli, 2 Kedi Yin, 8 Xi Ling, 3,9 Xinwei
Supporting Information Tuning Local Electronic Structure of Single Layer MoS2 through Defect Engineering Yan Chen, 1,2,,$, * Shengxi Huang, 3,6, Xiang Ji, 2 Kiran Adepalli, 2 Kedi Yin, 8 Xi Ling, 3,9 Xinwei
Supporting information. The Unusual and the Expected in the Si/C Phase Diagram. Guoying Gao, N. W. Ashcroft and Roald Hoffmann.
 Supporting information The Unusual and the Expected in the Si/C Phase Diagram Guoying Gao, N. W. Ashcroft and Roald Hoffmann Table of Contents Computational Methods...S1 Hypothetical Structures for Si
Supporting information The Unusual and the Expected in the Si/C Phase Diagram Guoying Gao, N. W. Ashcroft and Roald Hoffmann Table of Contents Computational Methods...S1 Hypothetical Structures for Si
Effect of magnetic disorder and strong electron correlations on the thermodynamics of CrN
 Effect of magnetic disorder and strong electron correlations on the thermodynamics of CrN Björn Alling, Tobias Marten and Igor Abrikosov Linköping University Post Print N.B.: When citing this work, cite
Effect of magnetic disorder and strong electron correlations on the thermodynamics of CrN Björn Alling, Tobias Marten and Igor Abrikosov Linköping University Post Print N.B.: When citing this work, cite
Exchange-induced negative-u charge order in N-doped WO 3 : A spin-peierls-like system
 Exchange-induced negative-u charge order in N-doped WO 3 : A spin-peierls-like system Muhammad N. Huda,*, Yanfa Yan, Su-Huai Wei, and Mowafak M. Al-Jassim National Renewable Energy Laboratory, Golden,
Exchange-induced negative-u charge order in N-doped WO 3 : A spin-peierls-like system Muhammad N. Huda,*, Yanfa Yan, Su-Huai Wei, and Mowafak M. Al-Jassim National Renewable Energy Laboratory, Golden,
University of Chinese Academy of Sciences, Beijing , People s Republic of China,
 SiC 2 Siligraphene and Nanotubes: Novel Donor Materials in Excitonic Solar Cell Liu-Jiang Zhou,, Yong-Fan Zhang, Li-Ming Wu *, State Key Laboratory of Structural Chemistry, Fujian Institute of Research
SiC 2 Siligraphene and Nanotubes: Novel Donor Materials in Excitonic Solar Cell Liu-Jiang Zhou,, Yong-Fan Zhang, Li-Ming Wu *, State Key Laboratory of Structural Chemistry, Fujian Institute of Research
Experiment Section Fig. S1 Fig. S2
 Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2018 Supplementary Materials Experiment Section The STM experiments were carried out in an ultrahigh
Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2018 Supplementary Materials Experiment Section The STM experiments were carried out in an ultrahigh
First-principles studies of the structural, electronic, and optical properties of a novel thorium compound Rb 2 Th 7 Se 15
 First-principles studies of the structural, electronic, and optical properties of a novel thorium compound Rb 2 Th 7 Se 15 M.G. Brik 1 Institute of Physics, University of Tartu, Riia 142, Tartu 5114, Estonia
First-principles studies of the structural, electronic, and optical properties of a novel thorium compound Rb 2 Th 7 Se 15 M.G. Brik 1 Institute of Physics, University of Tartu, Riia 142, Tartu 5114, Estonia
First-principles study of elastic properties of Cr-Al-N
 First-principles study of elastic properties of Cr-Al-N Liangcai Zhou, 1,* David Holec, 2 and Paul. H. Mayrhofer 1 1 Institute of Materials Science and Technology, Vienna University of Technology, A-1040
First-principles study of elastic properties of Cr-Al-N Liangcai Zhou, 1,* David Holec, 2 and Paul. H. Mayrhofer 1 1 Institute of Materials Science and Technology, Vienna University of Technology, A-1040
Role of N defects in paramagnetic CrN at finite temperatures from first principles
 Role of N defects in paramagnetic CrN at finite temperatures from first principles Elham Mozafari, Björn Alling, Peter Steneteg and Igor Abrikosov Linköping University Post Print N.B.: When citing this
Role of N defects in paramagnetic CrN at finite temperatures from first principles Elham Mozafari, Björn Alling, Peter Steneteg and Igor Abrikosov Linköping University Post Print N.B.: When citing this
Supporting Information for. Revealing Surface Elemental Composition and Dynamic Processes
 Supporting Information for Revealing Surface Elemental Composition and Dynamic Processes Involved in Facet-dependent Oxidation of Pt 3 Co Nanoparticles via in-situ Transmission Electron Microscopy Sheng
Supporting Information for Revealing Surface Elemental Composition and Dynamic Processes Involved in Facet-dependent Oxidation of Pt 3 Co Nanoparticles via in-situ Transmission Electron Microscopy Sheng
Explaining the apparent arbitrariness of the LDA-1/2 self-energy. correction method applied to purely covalent systems
 Explaining the apparent arbitrariness of the LDA-1/2 self-energy correction method applied to purely covalent systems Kan-Hao Xue, 1,2 Leonardo R. C. Fonseca, 3 and Xiang-Shui Miao 1,2 1 School of Optical
Explaining the apparent arbitrariness of the LDA-1/2 self-energy correction method applied to purely covalent systems Kan-Hao Xue, 1,2 Leonardo R. C. Fonseca, 3 and Xiang-Shui Miao 1,2 1 School of Optical
Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type
 Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type Zhigang Gu, a Lars Heinke, a,* Christof Wöll a, Tobias Neumann,
Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type Zhigang Gu, a Lars Heinke, a,* Christof Wöll a, Tobias Neumann,
arxiv:cond-mat/ v1 10 Jun 1994 K. M. Rabe
 October 2, 2018 Phase transitions in BaTiO 3 from first principles W. Zhong and David Vanderbilt Department of Physics and Astronomy, Rutgers University, Piscataway, NJ 08855-0849 arxiv:cond-mat/9406049v1
October 2, 2018 Phase transitions in BaTiO 3 from first principles W. Zhong and David Vanderbilt Department of Physics and Astronomy, Rutgers University, Piscataway, NJ 08855-0849 arxiv:cond-mat/9406049v1
Supporting Information for 2D Intrinsic. Ferromagnets from van der Waals. Antiferromagnets
 Supporting Information for 2D Intrinsic Ferromagnets from van der Waals Antiferromagnets Naihua Miao,, Bin Xu, Linggang Zhu,, Jian Zhou, and Zhimei Sun,, School of Materials Science and Engineering, Beihang
Supporting Information for 2D Intrinsic Ferromagnets from van der Waals Antiferromagnets Naihua Miao,, Bin Xu, Linggang Zhu,, Jian Zhou, and Zhimei Sun,, School of Materials Science and Engineering, Beihang
Design of Efficient Catalysts with Double Transition Metal. Atoms on C 2 N Layer
 Supporting Information Design of Efficient Catalysts with Double Transition Metal Atoms on C 2 N Layer Xiyu Li, 1, Wenhui Zhong, 2, Peng Cui, 1 Jun Li, 1 Jun Jiang 1, * 1 Hefei National Laboratory for
Supporting Information Design of Efficient Catalysts with Double Transition Metal Atoms on C 2 N Layer Xiyu Li, 1, Wenhui Zhong, 2, Peng Cui, 1 Jun Li, 1 Jun Jiang 1, * 1 Hefei National Laboratory for
Chromium Cluster on Defected Graphene
 Chromium Cluster on Defected Graphene Yuhang Liu June 29, 2017 Abstract In this work, diffusion process of Cr atoms on two types of defected graphene and structure and magnetic properties of Cr cluster
Chromium Cluster on Defected Graphene Yuhang Liu June 29, 2017 Abstract In this work, diffusion process of Cr atoms on two types of defected graphene and structure and magnetic properties of Cr cluster
Finite-temperature magnetism in bcc Fe under compression. Xianwei Sha* and R. E. Cohen
 Finite-temperature magnetism in bcc Fe under compression Xianwei Sha* and R. E. Cohen Carnegie Institution of Washington, 5251 Broad Branch Road, NW, Washington, D. C. 20015, U. S. A. We investigate the
Finite-temperature magnetism in bcc Fe under compression Xianwei Sha* and R. E. Cohen Carnegie Institution of Washington, 5251 Broad Branch Road, NW, Washington, D. C. 20015, U. S. A. We investigate the
EFFECTS OF STOICHIOMETRY ON POINT DEFECTS AND IMPURITIES IN GALLIUM NITRIDE
 EFFECTS OF STOICHIOMETRY ON POINT DEFECTS AND IMPURITIES IN GALLIUM NITRIDE C. G. VAN DE WALLE AND J. E. NORTHRUP Palo Alto Research Center, 3333 Coyote Hill Road, Palo Alto, CA 930, USA E-mail: vandewalle@parc.com
EFFECTS OF STOICHIOMETRY ON POINT DEFECTS AND IMPURITIES IN GALLIUM NITRIDE C. G. VAN DE WALLE AND J. E. NORTHRUP Palo Alto Research Center, 3333 Coyote Hill Road, Palo Alto, CA 930, USA E-mail: vandewalle@parc.com
On Dynamic and Elastic Stability of Lanthanum Carbide
 Journal of Physics: Conference Series On Dynamic and Elastic Stability of Lanthanum Carbide To cite this article: B D Sahoo et al 212 J. Phys.: Conf. Ser. 377 1287 Recent citations - Theoretical prediction
Journal of Physics: Conference Series On Dynamic and Elastic Stability of Lanthanum Carbide To cite this article: B D Sahoo et al 212 J. Phys.: Conf. Ser. 377 1287 Recent citations - Theoretical prediction
High Temperature High Pressure Properties of Silica From Quantum Monte Carlo
 High Temperature High Pressure Properties of Silica From Quantum Monte Carlo K.P. Driver, R.E. Cohen, Z. Wu, B. Militzer, P. Lopez Rios, M. Towler, R. Needs, and J.W. Wilkins Funding: NSF, DOE; Computation:
High Temperature High Pressure Properties of Silica From Quantum Monte Carlo K.P. Driver, R.E. Cohen, Z. Wu, B. Militzer, P. Lopez Rios, M. Towler, R. Needs, and J.W. Wilkins Funding: NSF, DOE; Computation:
2.1 Experimental and theoretical studies
 Chapter 2 NiO As stated before, the first-row transition-metal oxides are among the most interesting series of materials, exhibiting wide variations in physical properties related to electronic structure.
Chapter 2 NiO As stated before, the first-row transition-metal oxides are among the most interesting series of materials, exhibiting wide variations in physical properties related to electronic structure.
Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC
 286 Brazilian Journal of Physics, vol. 36, no. 2A, June, 2006 Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC Clas Persson and Susanne Mirbt Department
286 Brazilian Journal of Physics, vol. 36, no. 2A, June, 2006 Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC Clas Persson and Susanne Mirbt Department
Supporting Information. Don-Hyung Ha, Liane M. Moreau, Clive R. Bealing, Haitao Zhang, Richard G. Hennig, and. Richard D.
 Supporting Information The structural evolution and diffusion during the chemical transformation from cobalt to cobalt phosphide nanoparticles Don-Hyung Ha, Liane M. Moreau, Clive R. Bealing, Haitao Zhang,
Supporting Information The structural evolution and diffusion during the chemical transformation from cobalt to cobalt phosphide nanoparticles Don-Hyung Ha, Liane M. Moreau, Clive R. Bealing, Haitao Zhang,
STRUCTURAL AND MECHANICAL PROPERTIES OF AMORPHOUS SILICON: AB-INITIO AND CLASSICAL MOLECULAR DYNAMICS STUDY
 STRUCTURAL AND MECHANICAL PROPERTIES OF AMORPHOUS SILICON: AB-INITIO AND CLASSICAL MOLECULAR DYNAMICS STUDY S. Hara, T. Kumagai, S. Izumi and S. Sakai Department of mechanical engineering, University of
STRUCTURAL AND MECHANICAL PROPERTIES OF AMORPHOUS SILICON: AB-INITIO AND CLASSICAL MOLECULAR DYNAMICS STUDY S. Hara, T. Kumagai, S. Izumi and S. Sakai Department of mechanical engineering, University of
Supporting Information
 Supporting Information Controlled Growth of Ceria Nanoarrays on Anatase Titania Powder: A Bottom-up Physical Picture Hyun You Kim 1, Mark S. Hybertsen 2*, and Ping Liu 2* 1 Department of Materials Science
Supporting Information Controlled Growth of Ceria Nanoarrays on Anatase Titania Powder: A Bottom-up Physical Picture Hyun You Kim 1, Mark S. Hybertsen 2*, and Ping Liu 2* 1 Department of Materials Science
Supporting information. Origins of High Electrolyte-Electrode Interfacial Resistances in Lithium Cells. Containing Garnet Type LLZO Solid Electrolytes
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting information Origins of High Electrolyte-Electrode Interfacial Resistances
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting information Origins of High Electrolyte-Electrode Interfacial Resistances
Structure, energetics, and mechanical stability of Fe-Cu bcc alloys from first-principles calculations
 Structure, energetics, and mechanical stability of Fe-Cu bcc alloys from first-principles calculations Jefferson Z. Liu, A. van de Walle, G. Ghosh, and M. Asta* Department of Materials Science and Engineering,
Structure, energetics, and mechanical stability of Fe-Cu bcc alloys from first-principles calculations Jefferson Z. Liu, A. van de Walle, G. Ghosh, and M. Asta* Department of Materials Science and Engineering,
Surface stress and relaxation in metals
 J. Phys.: Condens. Matter 12 (2000) 5541 5550. Printed in the UK PII: S0953-8984(00)11386-4 Surface stress and relaxation in metals P M Marcus, Xianghong Qian and Wolfgang Hübner IBM Research Center, Yorktown
J. Phys.: Condens. Matter 12 (2000) 5541 5550. Printed in the UK PII: S0953-8984(00)11386-4 Surface stress and relaxation in metals P M Marcus, Xianghong Qian and Wolfgang Hübner IBM Research Center, Yorktown
Supporting Information
 Electronic Supplementary Material (ESI) for Energy & Environmental Science. This journal is The Royal Society of Chemistry 2015 Supporting Information Pyrite FeS 2 for High-rate and Long-life Rechargeable
Electronic Supplementary Material (ESI) for Energy & Environmental Science. This journal is The Royal Society of Chemistry 2015 Supporting Information Pyrite FeS 2 for High-rate and Long-life Rechargeable
HIGH-PRESSURE CHARACTERISTICS OF α Fe 2 O 3 USING DFT+U
 HIGH-PRESSURE CHARACTERISTICS OF α Fe 2 O 3 USING DFT+U G. ROLLMANN and P. ENTEL Institute of Physics, University of Duisburg-Essen, 47048 Duisburg, Germany A. ROHRBACH and J. HAFNER Center for Computational
HIGH-PRESSURE CHARACTERISTICS OF α Fe 2 O 3 USING DFT+U G. ROLLMANN and P. ENTEL Institute of Physics, University of Duisburg-Essen, 47048 Duisburg, Germany A. ROHRBACH and J. HAFNER Center for Computational
SUPPLEMENTARY INFORMATION
 Supplementary Methods Materials Synthesis The In 4 Se 3-δ crystal ingots were grown by the Bridgeman method. The In and Se elements were placed in an evacuated quartz ampoule with an excess of In (5-10
Supplementary Methods Materials Synthesis The In 4 Se 3-δ crystal ingots were grown by the Bridgeman method. The In and Se elements were placed in an evacuated quartz ampoule with an excess of In (5-10
Supporting Information for
 Supporting Information for Pb-activated Amine-assisted Photocatalytic Hydrogen Evolution Reaction on Organic-Inorganic Perovskites Lu Wang *,,, Hai Xiao, Tao Cheng, Youyong Li *,, William A. Goddard III
Supporting Information for Pb-activated Amine-assisted Photocatalytic Hydrogen Evolution Reaction on Organic-Inorganic Perovskites Lu Wang *,,, Hai Xiao, Tao Cheng, Youyong Li *,, William A. Goddard III
This is an author-deposited version published in: Eprints ID: URL:
 Open Archive Toulouse Archive Ouverte (OATAO) OATAO is an open access repository that collects the work of Toulouse researchers and makes it freely available over the web where possible. This is an author-deposited
Open Archive Toulouse Archive Ouverte (OATAO) OATAO is an open access repository that collects the work of Toulouse researchers and makes it freely available over the web where possible. This is an author-deposited
Supplementary Figure 1. HRTEM images of PtNi / Ni-B composite exposed to electron beam. The. scale bars are 5 nm.
 Supplementary Figure 1. HRTEM images of PtNi / Ni-B composite exposed to electron beam. The scale bars are 5 nm. S1 Supplementary Figure 2. TEM image of PtNi/Ni-B composite obtained under N 2 protection.
Supplementary Figure 1. HRTEM images of PtNi / Ni-B composite exposed to electron beam. The scale bars are 5 nm. S1 Supplementary Figure 2. TEM image of PtNi/Ni-B composite obtained under N 2 protection.
Two-Dimensional CH 3 NH 3 PbI 3 Perovskite: Synthesis and Optoelectronic Application
 Two-Dimensional CH 3 NH 3 PbI 3 Perovskite: Synthesis and Optoelectronic Application Jingying Liu,, Yunzhou Xue,,, Ziyu Wang,, Zai-Quan Xu, Changxi Zheng, Bent Weber, Jingchao Song, Yusheng Wang, Yuerui
Two-Dimensional CH 3 NH 3 PbI 3 Perovskite: Synthesis and Optoelectronic Application Jingying Liu,, Yunzhou Xue,,, Ziyu Wang,, Zai-Quan Xu, Changxi Zheng, Bent Weber, Jingchao Song, Yusheng Wang, Yuerui
Our first-principles calculations were performed using the Vienna Ab Initio Simulation
 Supplementary Note 1: Computational details First-principles calculations Our first-principles calculations were performed using the Vienna Ab Initio Simulation Package (VASP) 1, which is based on density
Supplementary Note 1: Computational details First-principles calculations Our first-principles calculations were performed using the Vienna Ab Initio Simulation Package (VASP) 1, which is based on density
First-principles studies of cation-doped spinel LiMn 2 O 4 for lithium ion batteries
 First-principles studies of cation-doped spinel LiMn 2 O 4 for lithium ion batteries Siqi Shi, 1 Ding-sheng Wang, 2 Sheng Meng, 2 Liquan Chen, 1 and Xuejie Huang 1, * 1 Nanoscale Physics and Devices Laboratory,
First-principles studies of cation-doped spinel LiMn 2 O 4 for lithium ion batteries Siqi Shi, 1 Ding-sheng Wang, 2 Sheng Meng, 2 Liquan Chen, 1 and Xuejie Huang 1, * 1 Nanoscale Physics and Devices Laboratory,
Crystalline and Magnetic Anisotropy of the 3d Transition-Metal Monoxides
 Crystalline and of the 3d Transition-Metal Monoxides Institut für Festkörpertheorie und -optik Friedrich-Schiller-Universität Max-Wien-Platz 1 07743 Jena 2012-03-23 Introduction Crystalline Anisotropy
Crystalline and of the 3d Transition-Metal Monoxides Institut für Festkörpertheorie und -optik Friedrich-Schiller-Universität Max-Wien-Platz 1 07743 Jena 2012-03-23 Introduction Crystalline Anisotropy
Effects of substitutions of C atoms by Al and N in the w-aln compound
 Journal of Physics: Conference Series PAPER OPEN ACCESS Effects of substitutions of C atoms by Al and N in the w-aln compound To cite this article: J F Murillo et al 2016 J. Phys.: Conf. Ser. 687 012114
Journal of Physics: Conference Series PAPER OPEN ACCESS Effects of substitutions of C atoms by Al and N in the w-aln compound To cite this article: J F Murillo et al 2016 J. Phys.: Conf. Ser. 687 012114
First Principles Calculation of Defect and Magnetic Structures in FeCo
 Materials Transactions, Vol. 47, No. 11 (26) pp. 2646 to 26 Special Issue on Advances in Computational Materials Science and Engineering IV #26 The Japan Institute of Metals First Principles Calculation
Materials Transactions, Vol. 47, No. 11 (26) pp. 2646 to 26 Special Issue on Advances in Computational Materials Science and Engineering IV #26 The Japan Institute of Metals First Principles Calculation
First-Principles Calculation of Point Defects in Uranium Dioxide
 Materials Transactions, Vol. 47, No. 11 (26) pp. 2651 to 2657 Special Issue on Advances in Computational Materials Science and Engineering IV #26 The Japan Institute of Metals First-Principles Calculation
Materials Transactions, Vol. 47, No. 11 (26) pp. 2651 to 2657 Special Issue on Advances in Computational Materials Science and Engineering IV #26 The Japan Institute of Metals First-Principles Calculation
Supplementary Information. Structural Transition and Unusually Strong Antiferromagnetic Superexchange Coupling in Perovskite KAgF3
 Supplementary Information Structural Transition and Unusually Strong Antiferromagnetic Superexchange Coupling in Perovskite KAgF3 Dominik Kurzydłowski, *a Zoran Mazej, b Zvonko Jagličić, c Yaroslav Filinchuk
Supplementary Information Structural Transition and Unusually Strong Antiferromagnetic Superexchange Coupling in Perovskite KAgF3 Dominik Kurzydłowski, *a Zoran Mazej, b Zvonko Jagličić, c Yaroslav Filinchuk
Electric field dependent sound velocity change in Ba 1 x Ca x TiO 3 ferroelectric perovskites
 Indian Journal of Pure & Applied Physics Vol. 49, February 2011, pp. 132-136 Electric field dependent sound velocity change in Ba 1 x Ca x TiO 3 ferroelectric perovskites Dushyant Pradeep, U C Naithani
Indian Journal of Pure & Applied Physics Vol. 49, February 2011, pp. 132-136 Electric field dependent sound velocity change in Ba 1 x Ca x TiO 3 ferroelectric perovskites Dushyant Pradeep, U C Naithani
Supporting Information
 Supporting Information A Porous Two-Dimensional Monolayer Metal-Organic Framework Material and its Use for the Size-Selective Separation of Nanoparticles Yi Jiang, 1 Gyeong Hee Ryu, 1, 3 Se Hun Joo, 4
Supporting Information A Porous Two-Dimensional Monolayer Metal-Organic Framework Material and its Use for the Size-Selective Separation of Nanoparticles Yi Jiang, 1 Gyeong Hee Ryu, 1, 3 Se Hun Joo, 4
FULL POTENTIAL LINEARIZED AUGMENTED PLANE WAVE (FP-LAPW) IN THE FRAMEWORK OF DENSITY FUNCTIONAL THEORY
 IN THE FRAMEWORK OF DENSITY FUNCTIONAL THEORY") FULL POTENTIAL LINEARIZED AUGMENTED PLANE WAVE (FP-LAPW) IN THE FRAMEWORK OF DENSITY FUNCTIONAL THEORY C.A. Madu and B.N Onwuagba Department of Physics, Federal University of Technology Owerri, Nigeria
FULL POTENTIAL LINEARIZED AUGMENTED PLANE WAVE (FP-LAPW) IN THE FRAMEWORK OF DENSITY FUNCTIONAL THEORY C.A. Madu and B.N Onwuagba Department of Physics, Federal University of Technology Owerri, Nigeria
Structure of CoO(001) surface from DFT+U calculations
 surface from DFT+U calculations") Structure of CoO(001) surface from DFT+U calculations B. Sitamtze Youmbi and F. Calvayrac Institut des Molécules et Matériaux du Mans (IMMM), UMR CNRS 6283 16 septembre 2013 Introduction Motivation Motivation
Structure of CoO(001) surface from DFT+U calculations B. Sitamtze Youmbi and F. Calvayrac Institut des Molécules et Matériaux du Mans (IMMM), UMR CNRS 6283 16 septembre 2013 Introduction Motivation Motivation
Author(s) Togo, Atsushi; Oba, Fumiyasu; Tanak. Citation PHYSICAL REVIEW B (2008), 78(13)
 Togo, Atsushi; Oba, Fumiyasu; Tanak. Citation PHYSICAL REVIEW B (2008), 78(13)") Title First-principles calculations of th between rutile-type and CaCl-type Author(s) Togo, Atsushi; Oba, Fumiyasu; Tanak Citation PHYSICAL REVIEW B (8), 78(3) Issue Date 8- URL http://hdl.handle.net/433/8463
Title First-principles calculations of th between rutile-type and CaCl-type Author(s) Togo, Atsushi; Oba, Fumiyasu; Tanak Citation PHYSICAL REVIEW B (8), 78(3) Issue Date 8- URL http://hdl.handle.net/433/8463
Mustafa Uludogan 1, Tahir Cagin, William A. Goddard, III Materials and Process Simulation Center, Caltech, Pasadena, CA 91125, U.S.A.
 Ab Initio Studies On Phase Behavior of Barium Titanate Mustafa Uludogan 1, Tahir Cagin, William A. Goddard, III Materials and Process Simulation Center, Caltech, Pasadena, CA 91125, U.S.A. 1 Physics Department,
Ab Initio Studies On Phase Behavior of Barium Titanate Mustafa Uludogan 1, Tahir Cagin, William A. Goddard, III Materials and Process Simulation Center, Caltech, Pasadena, CA 91125, U.S.A. 1 Physics Department,
Supporting Information. Potential semiconducting and superconducting metastable Si 3 C. structures under pressure
 Supporting Information Potential semiconducting and superconducting metastable Si 3 C structures under pressure Guoying Gao 1,3,* Xiaowei Liang, 1 Neil W. Ashcroft 2 and Roald Hoffmann 3,* 1 State Key
Supporting Information Potential semiconducting and superconducting metastable Si 3 C structures under pressure Guoying Gao 1,3,* Xiaowei Liang, 1 Neil W. Ashcroft 2 and Roald Hoffmann 3,* 1 State Key
Atomic Resolution Interfacial Structure of Lead-free Ferroelectric
 Atomic Resolution Interfacial Structure of Lead-free Ferroelectric K 0.5 Na 0.5 NbO 3 Thin films Deposited on SrTiO 3 Chao Li 1, Lingyan Wang 1*, Zhao Wang 2, Yaodong Yang 2, Wei Ren 1 and Guang Yang 1
Atomic Resolution Interfacial Structure of Lead-free Ferroelectric K 0.5 Na 0.5 NbO 3 Thin films Deposited on SrTiO 3 Chao Li 1, Lingyan Wang 1*, Zhao Wang 2, Yaodong Yang 2, Wei Ren 1 and Guang Yang 1
Chapter 3. The (L)APW+lo Method. 3.1 Choosing A Basis Set
APW+lo Method. 3.1 Choosing A Basis Set") Chapter 3 The (L)APW+lo Method 3.1 Choosing A Basis Set The Kohn-Sham equations (Eq. (2.17)) provide a formulation of how to practically find a solution to the Hohenberg-Kohn functional (Eq. (2.15)). Nevertheless
Chapter 3 The (L)APW+lo Method 3.1 Choosing A Basis Set The Kohn-Sham equations (Eq. (2.17)) provide a formulation of how to practically find a solution to the Hohenberg-Kohn functional (Eq. (2.15)). Nevertheless
TUTORIAL 6: PHONONS, LATTICE EXPANSION, AND BAND-GAP RENORMALIZATION
 Hands-On Tutorial Workshop, July 29 th 2014 TUTORIAL 6: PHONONS, LATTICE EXPANSION, AND BAND-GAP RENORMALIZATION Christian Carbogno & Manuel Schöttler Fritz-Haber-Institut der Max-Planck-Gesellschaft,
Hands-On Tutorial Workshop, July 29 th 2014 TUTORIAL 6: PHONONS, LATTICE EXPANSION, AND BAND-GAP RENORMALIZATION Christian Carbogno & Manuel Schöttler Fritz-Haber-Institut der Max-Planck-Gesellschaft,
1 IMEM-CNR, U.O.S. Genova, Via Dodecaneso 33, Genova, IT. 2 Dipartimento di Fisica, Università di Genova, Via Dodecaneso 33, Genova, IT
 Spontaneous Oxidation of Ni Nanoclusters on MgO Monolayers Induced by Segregation of Interfacial Oxygen. M. Smerieri 1, J. Pal 1,2, L. Savio 1*, L. Vattuone 1,2, R. Ferrando 1,3, S. Tosoni 4, L. Giordano
Spontaneous Oxidation of Ni Nanoclusters on MgO Monolayers Induced by Segregation of Interfacial Oxygen. M. Smerieri 1, J. Pal 1,2, L. Savio 1*, L. Vattuone 1,2, R. Ferrando 1,3, S. Tosoni 4, L. Giordano
Spin crossovers in the Earth mantle. Spin crossovers in the Earth mantle
 Spin crossovers in the Earth mantle Spin crossovers in the Earth mantle Renata M. Wentzcovitch Dept. of Chemical Engineering and Materials Science Minnesota Supercomputing Institute Collaborators Han Hsu
Spin crossovers in the Earth mantle Spin crossovers in the Earth mantle Renata M. Wentzcovitch Dept. of Chemical Engineering and Materials Science Minnesota Supercomputing Institute Collaborators Han Hsu
Colloque National sur les Techniques de Modélisation et de Simulation en Science des Matériaux, Sidi Bel-Abbès Novembre 2009
 Colloque National sur les Techniques de Modélisation et de Simulation en Science des Matériaux, Sidi Bel-Abbès. 23-24 Novembre 2009 Elastic, electronic and optical properties of SiGe 2N 4 under pressure
Colloque National sur les Techniques de Modélisation et de Simulation en Science des Matériaux, Sidi Bel-Abbès. 23-24 Novembre 2009 Elastic, electronic and optical properties of SiGe 2N 4 under pressure
* Theoretische Physik II, Universitat Dortmund, Dortmund, Germany
 JOURNAL DE PHYSIQUE IV Colloque C8, suppl6ment au Journal de Physique III, Volume 5, dkembre 1995 Structural Phase Transformation and Phonon Softening in Iron-Based Alloys H.C. Herper, E. Hoffmann, P.
JOURNAL DE PHYSIQUE IV Colloque C8, suppl6ment au Journal de Physique III, Volume 5, dkembre 1995 Structural Phase Transformation and Phonon Softening in Iron-Based Alloys H.C. Herper, E. Hoffmann, P.
The Gutzwiller Density Functional Theory
 The Gutzwiller Density Functional Theory Jörg Bünemann, BTU Cottbus I) Introduction 1. Model for an H 2 -molecule 2. Transition metals and their compounds II) Gutzwiller variational theory 1. Gutzwiller
The Gutzwiller Density Functional Theory Jörg Bünemann, BTU Cottbus I) Introduction 1. Model for an H 2 -molecule 2. Transition metals and their compounds II) Gutzwiller variational theory 1. Gutzwiller
Effect of interfacial dislocations on ferroelectric phase stability and domain morphology in a thin film a phase-field model
 JOURNAL OF APPLIED PHYSICS VOLUME 94, NUMBER 4 15 AUGUST 2003 Effect of interfacial dislocations on ferroelectric phase stability and domain morphology in a thin film a phase-field model S. Y. Hu, Y. L.
JOURNAL OF APPLIED PHYSICS VOLUME 94, NUMBER 4 15 AUGUST 2003 Effect of interfacial dislocations on ferroelectric phase stability and domain morphology in a thin film a phase-field model S. Y. Hu, Y. L.
arxiv: v1 [cond-mat.mtrl-sci] 14 Apr 2010
![arxiv: v1 [cond-mat.mtrl-sci] 14 Apr 2010](/thumbs/72/66735818.jpg "arxiv: v1 [cond-mat.mtrl-sci] 14 Apr 2010") APS/123-QED Theory of the ferromagnetism in Ti 1 x Cr x N solid solutions B. Alling Department of Physics, Chemistry and Biology (IFM), arxiv:1004.2337v1 [cond-mat.mtrl-sci] 14 Apr 2010 Linköping University,
APS/123-QED Theory of the ferromagnetism in Ti 1 x Cr x N solid solutions B. Alling Department of Physics, Chemistry and Biology (IFM), arxiv:1004.2337v1 [cond-mat.mtrl-sci] 14 Apr 2010 Linköping University,
ELECTRONIC AND MAGNETIC PROPERTIES OF BERKELIUM MONONITRIDE BKN: A FIRST- PRINCIPLES STUDY
 ELECTRONIC AND MAGNETIC PROPERTIES OF BERKELIUM MONONITRIDE BKN: A FIRST- PRINCIPLES STUDY Gitanjali Pagare Department of Physics, Sarojini Naidu Govt. Girls P. G. Auto. College, Bhopal ( India) ABSTRACT
ELECTRONIC AND MAGNETIC PROPERTIES OF BERKELIUM MONONITRIDE BKN: A FIRST- PRINCIPLES STUDY Gitanjali Pagare Department of Physics, Sarojini Naidu Govt. Girls P. G. Auto. College, Bhopal ( India) ABSTRACT
Supporting Information
 Supporting Information The Origin of Active Oxygen in a Ternary CuO x /Co 3 O 4 -CeO Catalyst for CO Oxidation Zhigang Liu, *, Zili Wu, *, Xihong Peng, ++ Andrew Binder, Songhai Chai, Sheng Dai *,, School
Supporting Information The Origin of Active Oxygen in a Ternary CuO x /Co 3 O 4 -CeO Catalyst for CO Oxidation Zhigang Liu, *, Zili Wu, *, Xihong Peng, ++ Andrew Binder, Songhai Chai, Sheng Dai *,, School
Puckering and spin orbit interaction in nano-slabs
 Electronic structure of monolayers of group V atoms: Puckering and spin orbit interaction in nano-slabs Dat T. Do* and Subhendra D. Mahanti* Department of Physics and Astronomy, Michigan State University,
Electronic structure of monolayers of group V atoms: Puckering and spin orbit interaction in nano-slabs Dat T. Do* and Subhendra D. Mahanti* Department of Physics and Astronomy, Michigan State University,
Electronic, vibrational and thermodynamic properties of Ca 10 (AsO 4 ) 6 (OH) 2 : first principles study
 6 (OH) 2 : first principles study") Eur. Phys. J. Appl. Phys. (2015) 72: 31201 DOI: 10.1051/epjap/2015150301 Regular Article THE EUROPEAN PHYSICAL JOURNAL APPLIED PHYSICS Electronic, vibrational and thermodynamic properties of Ca 10 (AsO
Eur. Phys. J. Appl. Phys. (2015) 72: 31201 DOI: 10.1051/epjap/2015150301 Regular Article THE EUROPEAN PHYSICAL JOURNAL APPLIED PHYSICS Electronic, vibrational and thermodynamic properties of Ca 10 (AsO
Calculation and Analysis of the Dielectric Functions for BaTiO 3, PbTiO 3, and PbZrO 3
 CHINESE JOURNAL OF PHYSICS VOL. 1, NO. 3 June 213 Calculation and Analysis of the Dielectric Functions for BaTiO 3, PbTiO 3, and PbZrO 3 Chao Zhang and Dashu Yu School of Physics & Electronic Information
CHINESE JOURNAL OF PHYSICS VOL. 1, NO. 3 June 213 Calculation and Analysis of the Dielectric Functions for BaTiO 3, PbTiO 3, and PbZrO 3 Chao Zhang and Dashu Yu School of Physics & Electronic Information
Ab Initio Study of the Mechanical, Electronic, Thermal and Optical Properties of Ge 2 Sb 2 Te 5
 Ab Initio Study of the Mechanical, Electronic, Thermal and Optical Properties of Ge 2 Sb 2 Te 5 Odhiambo H. 1, Amolo G. 2, Makau N. 2, Othieno H. 1, and Oduor A. 1 1 Department of Physics, Maseno University,
Ab Initio Study of the Mechanical, Electronic, Thermal and Optical Properties of Ge 2 Sb 2 Te 5 Odhiambo H. 1, Amolo G. 2, Makau N. 2, Othieno H. 1, and Oduor A. 1 1 Department of Physics, Maseno University,
PBS: FROM SOLIDS TO CLUSTERS
 PBS: FROM SOLIDS TO CLUSTERS E. HOFFMANN AND P. ENTEL Theoretische Tieftemperaturphysik Gerhard-Mercator-Universität Duisburg, Lotharstraße 1 47048 Duisburg, Germany Semiconducting nanocrystallites like
PBS: FROM SOLIDS TO CLUSTERS E. HOFFMANN AND P. ENTEL Theoretische Tieftemperaturphysik Gerhard-Mercator-Universität Duisburg, Lotharstraße 1 47048 Duisburg, Germany Semiconducting nanocrystallites like
Teoría del Funcional de la Densidad (Density Functional Theory)
") Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Supporting Information
 Electronic Supplementary Material (ESI) for Energy & Environmental Science. This journal is The Royal Society of Chemistry 2017 Supporting Information Large Enhancement of Thermoelectric Properties in
Electronic Supplementary Material (ESI) for Energy & Environmental Science. This journal is The Royal Society of Chemistry 2017 Supporting Information Large Enhancement of Thermoelectric Properties in
Hydrogenation of Penta-Graphene Leads to Unexpected Large. Improvement in Thermal Conductivity
 Supplementary information for Hydrogenation of Penta-Graphene Leads to Unexpected Large Improvement in Thermal Conductivity Xufei Wu, a Vikas Varshney, b,c Jonghoon Lee, b,c Teng Zhang, a Jennifer L. Wohlwend,
Supplementary information for Hydrogenation of Penta-Graphene Leads to Unexpected Large Improvement in Thermal Conductivity Xufei Wu, a Vikas Varshney, b,c Jonghoon Lee, b,c Teng Zhang, a Jennifer L. Wohlwend,
Re-evaluating CeO 2 Expansion Upon Reduction: Non-counterpoised Forces, Not Ionic Radius Effects, are the Cause
 Re-evaluating CeO 2 Expansion Upon Reduction: Non-counterpoised Forces, Not Ionic Radius Effects, are the Cause Christopher L. Muhich, a* a ETH Zurich, Department of Mechanical and Process Engineering,
Re-evaluating CeO 2 Expansion Upon Reduction: Non-counterpoised Forces, Not Ionic Radius Effects, are the Cause Christopher L. Muhich, a* a ETH Zurich, Department of Mechanical and Process Engineering,
arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 14 May 2003
![arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 14 May 2003](/thumbs/75/72747568.jpg "arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 14 May 2003") LA-UR-3-239 arxiv:cond-mat/35331v1 [cond-mat.mtrl-sci] 14 May 23 Thermal Stabilization of the HCP Phase in Titanium Sven P. Rudin 1, M. D. Jones 2, and R. C. Albers 1 1 Los Alamos National Laboratory,
LA-UR-3-239 arxiv:cond-mat/35331v1 [cond-mat.mtrl-sci] 14 May 23 Thermal Stabilization of the HCP Phase in Titanium Sven P. Rudin 1, M. D. Jones 2, and R. C. Albers 1 1 Los Alamos National Laboratory,
First-principles calculations of structural, electronic and optical properties of HfZn 2
 ~ 1 ~ First-principles calculations of structural, electronic and optical properties of HfZn 2 Md. Atikur Rahman *1, Md. Afjalur Rahman 2, Md. Zahidur Rahaman 3 1, 2, 3 Department of Physics, Pabna University
~ 1 ~ First-principles calculations of structural, electronic and optical properties of HfZn 2 Md. Atikur Rahman *1, Md. Afjalur Rahman 2, Md. Zahidur Rahaman 3 1, 2, 3 Department of Physics, Pabna University
Supplementary Figures
 Supplementary Figures 8 6 Energy (ev 4 2 2 4 Γ M K Γ Supplementary Figure : Energy bands of antimonene along a high-symmetry path in the Brillouin zone, including spin-orbit coupling effects. Empty circles
Supplementary Figures 8 6 Energy (ev 4 2 2 4 Γ M K Γ Supplementary Figure : Energy bands of antimonene along a high-symmetry path in the Brillouin zone, including spin-orbit coupling effects. Empty circles
Optical properties of chalcopyrite-type intermediate transition metal band materials from first principles
 Optical properties of chalcopyrite-type intermediate transition metal band materials from first principles I. Aguilera, P. Palacios, P. Wahnon Institute de Energia Solar and Departamiento de Tecnologias
Optical properties of chalcopyrite-type intermediate transition metal band materials from first principles I. Aguilera, P. Palacios, P. Wahnon Institute de Energia Solar and Departamiento de Tecnologias
SCIENCE CHINA Physics, Mechanics & Astronomy. Electronic structure and optical properties of N-Zn co-doped -Ga 2 O 3
 SCIENCE CHINA Physics, Mechanics & Astronomy Article April 2012 Vol.55 No.4: 654 659 doi: 10.1007/s11433-012-4686-9 Electronic structure and optical properties of N-Zn co-doped -Ga 2 O 3 YAN JinLiang *
SCIENCE CHINA Physics, Mechanics & Astronomy Article April 2012 Vol.55 No.4: 654 659 doi: 10.1007/s11433-012-4686-9 Electronic structure and optical properties of N-Zn co-doped -Ga 2 O 3 YAN JinLiang *
Supporting Information
 Copyright WILEY-VCH Verlag GmbH & Co. KGaA, 69469 Weinheim, Germany, 2018. Supporting Information for Adv. Energy Mater., DOI: 10.1002/aenm.201800144 H 2 V 3 O 8 Nanowire/Graphene Electrodes for Aqueous
Copyright WILEY-VCH Verlag GmbH & Co. KGaA, 69469 Weinheim, Germany, 2018. Supporting Information for Adv. Energy Mater., DOI: 10.1002/aenm.201800144 H 2 V 3 O 8 Nanowire/Graphene Electrodes for Aqueous
Electronic and Vibrational Properties of Pbsns 3
 IOSR Journal of Electrical and Electronics Engineering (IOSR-JEEE) e-issn: 2278-1676,p-ISSN: 2320-3331, Volume 5, Issue 5 (May. - Jun. 2013), PP 12-17 N. N. Omehe 1, S. Ehika 2 and S. O. Azi 3 1 Federal
IOSR Journal of Electrical and Electronics Engineering (IOSR-JEEE) e-issn: 2278-1676,p-ISSN: 2320-3331, Volume 5, Issue 5 (May. - Jun. 2013), PP 12-17 N. N. Omehe 1, S. Ehika 2 and S. O. Azi 3 1 Federal
ab initio Lattice Vibrations: Calculating the Thermal Expansion Coeffcient Felix Hanke & Martin Fuchs June 30, 2009 This afternoon s plan
 ab initio Lattice Vibrations: Calculating the Thermal Expansion Coeffcient Felix Hanke & Martin Fuchs June 3, 29 This afternoon s plan introductory talk Phonons: harmonic vibrations for solids Phonons:
ab initio Lattice Vibrations: Calculating the Thermal Expansion Coeffcient Felix Hanke & Martin Fuchs June 3, 29 This afternoon s plan introductory talk Phonons: harmonic vibrations for solids Phonons:
TUTORIAL 8: PHONONS, LATTICE EXPANSION, AND BAND-GAP RENORMALIZATION
 TUTORIAL 8: PHONONS, LATTICE EXPANSION, AND BAND-GAP RENORMALIZATION 1 INVESTIGATED SYSTEM: Silicon, diamond structure Electronic and 0K properties see W. Huhn, Tutorial 2, Wednesday August 2 2 THE HARMONIC
TUTORIAL 8: PHONONS, LATTICE EXPANSION, AND BAND-GAP RENORMALIZATION 1 INVESTIGATED SYSTEM: Silicon, diamond structure Electronic and 0K properties see W. Huhn, Tutorial 2, Wednesday August 2 2 THE HARMONIC
Free energy sampling for electrochemical systems
 Free energy sampling for electrochemical systems Mira Todorova, Anoop Kishore Vatti, Suhyun Yoo and Jörg Neugebauer Department of Computational Materials Design Düsseldorf, Germany m.todorova@mpie.de IPAM,
Free energy sampling for electrochemical systems Mira Todorova, Anoop Kishore Vatti, Suhyun Yoo and Jörg Neugebauer Department of Computational Materials Design Düsseldorf, Germany m.todorova@mpie.de IPAM,
UTC Power, South Windsor, CT United Technologies Research Center, East Hartford, CT
 Supporting Information Electrocatalysis on Platinum Nanoparticles: Particle Size Effect on Oxygen Reduction Reaction Activity Minhua Shao,, * Amra Peles,, * Krista Shoemaker UTC Power, South Windsor, CT
Supporting Information Electrocatalysis on Platinum Nanoparticles: Particle Size Effect on Oxygen Reduction Reaction Activity Minhua Shao,, * Amra Peles,, * Krista Shoemaker UTC Power, South Windsor, CT
arxiv: v1 [cond-mat.mtrl-sci] 18 Sep 2013
![arxiv: v1 [cond-mat.mtrl-sci] 18 Sep 2013](/thumbs/79/80409317.jpg "arxiv: v1 [cond-mat.mtrl-sci] 18 Sep 2013") Accuracy of generalized gradient approximation functionals for density functional perturbation theory calculations arxiv:1309.4805v1 [cond-mat.mtrl-sci] 18 Sep 2013 Lianhua He, 1 Fang Liu, 2 Geoffroy Hautier,
Accuracy of generalized gradient approximation functionals for density functional perturbation theory calculations arxiv:1309.4805v1 [cond-mat.mtrl-sci] 18 Sep 2013 Lianhua He, 1 Fang Liu, 2 Geoffroy Hautier,
Southeast University, Nanjing, China 2 Department of Applied Physics, Aalto University,
 Supplementary Information to Solubility of Boron, Carbon and Nitrogen in Transition Metals: Getting Insight into Trends from First-Principles Calculations Xiaohui Hu, 1,2 Torbjörn Björkman 2,3, Harri Lipsanen
Supplementary Information to Solubility of Boron, Carbon and Nitrogen in Transition Metals: Getting Insight into Trends from First-Principles Calculations Xiaohui Hu, 1,2 Torbjörn Björkman 2,3, Harri Lipsanen
Phonons In The Elk Code
 Phonons In The Elk Code Kay Dewhurst, Sangeeta Sharma, Antonio Sanna, Hardy Gross Max-Planck-Institut für Mikrostrukturphysik, Halle If you are allowed to measure only one property of a material, make
Phonons In The Elk Code Kay Dewhurst, Sangeeta Sharma, Antonio Sanna, Hardy Gross Max-Planck-Institut für Mikrostrukturphysik, Halle If you are allowed to measure only one property of a material, make
Outline. Introduction: graphene. Adsorption on graphene: - Chemisorption - Physisorption. Summary
 Outline Introduction: graphene Adsorption on graphene: - Chemisorption - Physisorption Summary 1 Electronic band structure: Electronic properties K Γ M v F = 10 6 ms -1 = c/300 massless Dirac particles!
Outline Introduction: graphene Adsorption on graphene: - Chemisorption - Physisorption Summary 1 Electronic band structure: Electronic properties K Γ M v F = 10 6 ms -1 = c/300 massless Dirac particles!