Structural and Electronic Effects on the Properties of Fe 2 (dobdc) upon Oxidation with N 2 O
|
|
|
- Laurel Craig
- 6 years ago
- Views:
Transcription
1 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-1 Structural and Electronic Effects on the Properties of Fe (dobdc) upon Oxidation with N O oshua Borycz, 1, oachim Paier, 3,* Pragya Verma, 1, Lucy E. Darago,,4 Dianne. Xiao,,4 Donald G. Truhlar, 1,, * effrey R. Long,,4-6 and Laura Gagliardi 1,, * 1 Department of Chemistry, Minnesota Supercomputing Institute, and Chemical Theory Center, University of Minnesota, 07 Pleasant Street SE, Minneapolis, Minnesota , USA. Nanoporous Materials Genome Center, University of Minnesota, 07 Pleasant Street SE, Minneapolis, Minnesota , USA. 3 Institut für Chemie, Humboldt-Universität zu Berlin, Unter den Linden 6, Berlin, Germany. 4 Department of Chemistry, University of California, Berkeley, California , USA. Department of Chemical and Biomolecular Engineering and Chemistry, University of California, Berkeley, California , USA. 6 Materials Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 9470, USA. * To whom the correspondence should be addressed: gagliardi@umn.edu, joachim.paier@chemie.hu-berlin.de, truhlar@umn.edu
2 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S- Table of Contents 1. Lattice Parameters S-. Equilibrium Structure Comparison S-3 3. Convergence Tests and Grids S-4 4. Elastic Properties S-6. Infrared Spectra S-8 6. Orbital Projected Density of States (OP-DOS) S-9 7. Magnetic constants computed assuming z = 3 S Doubled Unit Cell Calculations S Cluster Calculations S Optimized Periodic Structures (VASP Format) S- 11. Cluster Model Structures (xyz Format) S-4 1. Charges and spin populations S Curie-Weiss Fit S References S-9 1. Lattice Parameters Table S1: Lattice parameters for Fe (dobdc), Fe (O) (dobdc), and Fe (OH) (dobdc). method a (Å) b (Å) c (Å) α ( ) β ( ) γ ( ) volume (Å 3 ) Fe (dobdc) PBE PBE+U PBE+U+D HSE HSE+D Expt. a Fe (O) (dobdc) PBE PBE+U PBE+U+D HSE HSE+D Fe (OH) (dobdc) PBE PBE+U PBE+U+D HSE HSE+D Expt. a a Taken from ref 1.
3 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-3. Equilibrium Structure Comparison Table S gives the equilibrium structures of the three MOFs studied here. In the case of Fe (dobdc) there is good agreement between the bond distances and angles computed with all the methods and experiment, with the exception of PBE, which significantly underestimates the Fe Fe distances and the Fe O c Fe angle. These findings are expected based on previous calculations with PBE and Fe (dobdc)., 3 The good agreement of GAM+U, PBE+U, and HSE06 for Fe (dobdc) is quite encouraging. Table S shows that after oxidation to Fe(III), the experimental Fe Fe distance increases by Å. If we ignore PBE due its clear underestimation of the Fe Fe distance in Fe (dobdc), the density functional calculations predict that there is an increase in the Fe Fe distance between 0. and 0.8 Å. This consistency indicates that the geometrical parameters are predicted reasonably accurately. The comparison of theory and experiment may be complicated by the presence of Fe(II) in the experimental Fe (OH) (dobdc) sample, leading to structural disorder of the Fe(II) and Fe(III) sites. The discrepancies may also be the result of the structural averaging that takes place when performing refinements from X-ray powder diffraction data. For the Fe(IV) case, the various calculations are in good agreement with one another, but there is no experimental data for comparison. The only structural parameter that is improved significantly by including Hartree Fock exchange is the unit cell volume (Table S1). Changes in the equilibrium volumes are small upon inclusion of molecular mechanics damped dispersion ( 0.% to 1%). This is reasonable based on the strong covalent bonds making up these MOFs. Volumes obtained using HSE06 are approximately % to 3% smaller than the volumes obtained using PBE+U.
4 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-4 Table S: Bond distances and angles for the nearest-neighbor iron and oxygen atoms in Fe (dobdc), Fe (O) (dobdc), and Fe (OH) (dobdc) computed using periodic DFT. All geometries presented were optimized in the FM spin state. Fe Fe Fe O Fe O a1 Fe O a Fe O b Fe O c1 Fe O c Fe O a Fe Fe O c Fe method length (Å) angle (deg.) Fe(II) case: Fe (dobdc) PBE PBE+U PBE+U-D HSE HSE06-D GAM+U Expt. (PXRD) a Fe(III) case: Fe (OH) (dobdc) PBE PBE+U PBE+U-D HSE HSE06-D GAM+U Expt.(PXRD) b Expt.(EXAFS) b c Fe(IV) case: Fe (O) (dobdc) PBE PBE+U PBE+U-D HSE HSE06-D GAM+U a ref 1, 4 b ref 1 c Average equatorial Fe-O distance 3. Convergence Tests and Grids For the PBE+U calculations, Table S3 reports our findings showing that the energies and structural parameters of Fe (dobdc) are well converged using a x x 4 k-point grid. For the HSE06 calculations, we did a number of separate tests to check convergence of the energy with respect to the number of k-points used to sample the Fock potential. To reduce the computational cost we tested the effect of reducing the k-point grid for the Fock potential to 1 x 1 x. 6 For Fe (dobdc), this resulted in a total energy change of 0.08 ev in the ferromagnetically (FM) ordered case. For the antiferromagnetically (AFM) ordered Fe (O) (dobdc) we found a change in energy of only ev with a 1 x 1 x k-point mesh. Thus, the HSE06 calculations employ the full k-point mesh for Fe (dobdc) and a reduced mesh for Fe (O) (dobdc) and Fe (OH) (dobdc). The grid reduction was not performed for Fe (dobdc) because it has unique metamagnetic character 7 that requires a dense k-point grid to
5 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S- be modeled accurately. For the density of states (DOS) calculations the number of grid points was increased to 001 with the NEDOS tag in VASP, and single-points were performed on the optimized PBE+U and HSE geometries. Table S3: Convergence of total energies and structural parameters of Fe (dobdc) with respect to the number of k-points to sample the BZ. Method: Dudarev s 8 DFT+U (PBE+U, U = ev; energy cut-off = 00 ev; ISIF = 3). #k points E 0 /UC [ev] a [Å] b [Å] c [Å] α [ ] β [ ] γ [ ] V UC [Å 3 ] 1x1x xx xx x4x
6 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-6 4. Elastic Properties 4.1. Methods The bulk modulus measures the response of a crystal to uniform pressure, i.e. the hardness of the material. The bulk moduli of each species were computed with PBE+U by incrementally expanding and shrinking the unit cell and optimizing the ionic positions. A total of eleven points were used for each volume scan (±30% of the equilibrium volume), and the computed curves were used to fit the Birch-Murnaghan equation of state (BM-EOS). 9 The bulk modulus and third-order BM-EOS are defined by the equations, = = (S1) (S) where V is the deformed volume, V 0 is the equilibrium volume, P is pressure, and B 0 is the first derivative of the bulk modulus with respect to pressure. The BM-EOS is commonly used to compute the bulk moduli of MOFs, and can provide a reasonable estimate of the relative hardness of the materials studied as well as an indication of the accuracy of the computed unit cell volumes, because it provides an estimation of the effect of anharmonicity. 4.. Results The curves used to predict the bulk moduli of the studied species are provided in Figure S1, and the ground state unit cell volumes and bulk moduli are presented in Table S4. The BM- EOS and PBE+U computed unit cell volumes are within Å 3 ( 0.4%), i.e. a perfect match, in the case of Fe (dobdc). These volumes are within 0 Å 3 ( +1.%) for Fe (O) (dobdc) and Fe (OH) (dobdc). Volumes obtained using BM-EOS for Fe (O) (dobdc) and Fe (OH) (dobdc) are larger than the volumes optimized using the stress tensor, and hence indicate some degree of anharmonicity in the E(V) curve shown in Figure S1. Since the bulk modulus is a measure of the pressure needed to change the unit cell volume, it was expected that adding O and OH to Fe (dobdc) would result in larger bulk moduli values.
7 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-7 Figure S1: Bulk moduli of Fe (dobdc) and Fe (OH) (dobdc) computed using PBE+U and using the BM-EOS. Note that Fe (O) (dobdc) curves are not shown for clarity. The energy of the equilibrium geometry was set to zero. Table S4: Minimum volumes and bulk moduli of Fe (dobdc), Fe (O) (dobdc), and Fe (OH) (dobdc) as predicted by PBE+U and by the BM-EOS. VASP V 0 (Å 3 ) BM V 0 (Å 3 ) B 0 (GPa) Fe (dobdc) Fe (O) (dobdc) Fe (OH) (dobdc)
8 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-8. Infrared Spectra Figure S: Above are the experimental and theoretical infrared spectra of Fe (dobdc) and below are the same for Fe (OH) (dobdc). All spectra were normalized based on the highest peak. The experimental spectra were taken from ref 1. To test the effect that freezing the carbon atoms has on the IR spectra, each plot shows the spectrum resulting from the vibrations of Fe and O and that arising from the vibrations of Fe, O, and Ca. The C a O stretches seem to have a negligible effect on the Fe O stretches and bends. For both Fe (dobdc) and Fe (OH) (dobdc) there are two C O peaks that are red-shifted from experiment by approximately 1 cm 1. When C a was unfrozen these peaks shifted towards the organic C O region and a new C C stretch appeared in the aromatic region).
 Density of States (OP-DOS) The OP-DOS comparison between Fe")


 Fe (OH) (dobdc) in their respective ground spin state.")
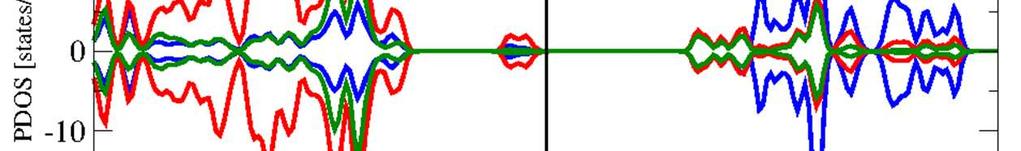
9 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-9 6. Orbital Projected (Local) Density of States (OP-DOS) The OP-DOS comparison between Fe (dobdc), Fe (O) (dobdc), and Fe (OH) (dobdc) at the PBE+U level is provided in Figure S3. The same comparison with HSE06 is provided in Figure S4. Figure S shows the projection of an (Fe O) unit in both Fe (dobdc) and Fe (O) (dobdc). Figure S3. PBE+U orbital projected densities of states of (a) Fe (dobdc), (b) Fe (O) (dobdc), and (c) Fe (OH) (dobdc) in their respective ground spin state. Densities given as positive are those for the majority spin, and densities shown as negative are those for the minority spin.

, (b) Fe (O) (dobdc), and (c) Fe (OH) (dobdc) in")


10 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-10 Figure S4. HSE06 orbital projected densities of states of (a) Fe (dobdc), (b) Fe (O) (dobdc), and (c) Fe (OH) (dobdc) in their respective ground spin states (see Table 1). Densities given as positive are those for the majority spin, and densities shown as negative are those for the minority spin. Figure S: OP-DOS of an (Fe O) unit in Fe (dobdc) (upper panel) and Fe (O) (dobdc) (lower panel).


11 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-11 By virtue of the very good agreement between PBE+U and HSE06 structural parameters, we found that it is efficient to simply rescale the volume by a factor of 0.97 in total, in particulr by 0.99 in each of the spatial directions, and do single-point HSE06 calculations on top of the rescaled PBE+U structures to compute the HSE06 magnetic coupling parameters. DOS obtained using this protocol agree well with the OP-DOS obtained by the more computationally expensive full optimizations using HSE06. The Fe (dobdc) OP- DOS for the ferromagnetic (FM), intrachain antiferromagnetic (AFM1), and interchain antiferromagnetic (AFM) spin states with both PBE+U and an HSE single-point upon scaling to PBE+U volume to the HSE optimized volume are provided in Figures S6-S8. The same quantities for Fe (O) (dobdc) and Fe (OH) (dobdc) are provided in Figures S9-S11 and S1-S14, respectively. Figure S1 shows a comparison of the Fe (dobdc), HSE OP-DOS with the HSE OP-DOS of the PBE+U geometry scaled to the HSE unit cell volume. Figure S16 shows the same for Fe (O) (dobdc). Figure S6: FM-ordered Fe (dobdc) OP-DOS.





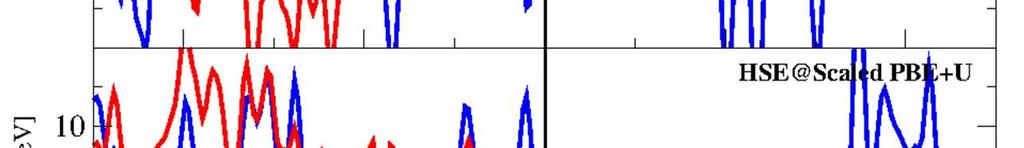
12 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-1 Figure S7: AFM1-ordered Fe (dobdc) OP-DOS. Figure S8: AFM-ordered Fe (dobdc) OP-DOS.



")

13 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-13 Figure S9: FM-ordered Fe (O) (dobdc) OP-DOS. Figure S10: AFM1-ordered Fe (O) (dobdc) OP-DOS.




14 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-14 Figure S11: AFM-ordered Fe (O) (dobdc) OP-DOS. Figure S1: FM-ordered Fe (OH) (dobdc) OP-DOS.


 (dobdc) OP-DOS.")
:")
15 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-1 Figure S13: AFM1-ordered Fe (OH) (dobdc) OP-DOS. Figure S14: AFM-ordered Fe (OH) (dobdc) OP-DOS. Fe (dobdc):
 of Fe")

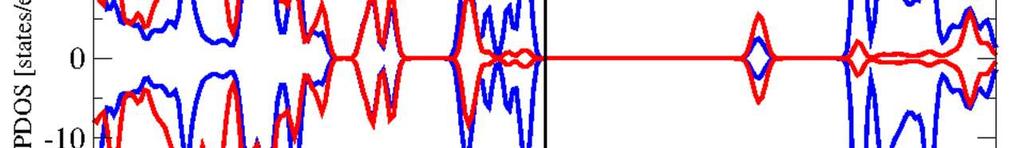
16 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-16 Figure S1: Upper panel: HSE OP-DOS at the corresponding fully optimized structure (+ volume) of Fe (dobdc) in FM ordering. Lower panel: HSE OP-DOS performed on PBE+U optimized coordinates that were scaled to the HSE unit cell volume. Fe (O) (dobdc) Figure S16: Upper panel: HSE OP-DOS at the corresponding fully optimized structure (+ volume) of Fe (O) (dobdc) in AFM ordering. Lower panel: HSE OP-DOS performed on PBE+U optimized coordinates that were scaled to the HSE unit cell volume.
17 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S Magnetic coupling values computed assuming z = 3 Table S: Periodic DFT isotropic exchange and coupling energies of the iron centers for each studied MOF. The NN and IC coupling values represent the nearest-neighbor and the interchain metal-metal couplings, respectively. All coupling values were extracted using geometries optimized with the FM spin state. method E AFM1 E b FM / b E AFM E FM NN ( NNN ) z IC b E AFM3 E FM cm 1 Fe(II) case: Fe (dobdc) PBE PBE+U-108 b 6.6/ (0.3) 1.9 PBE+U PBE+U-D HSE HSE06-D Expt. a Fe(III) case: Fe (OH) (dobdc) PBE PBE+U-13 b 316.0/ ( 1.1) 6.3 PBE+U PBE+U-D HSE HSE06-D Fe(IV) case: Fe (O) (dobdc) PBE PBE+U-10 b 0.6/ ( 0.9) 1.3 PBE+U PBE+U-D HSE HSE06-D a Taken from ref 7. b E FM is the electronic energy of the ferromagnetic state, E AFM1 is the electronic energy of antiferromagnetic state 1, and E AFM is the electronic energy of antiferromagnetic state.
18 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S Doubled Unit Cell Calculations Figure S17 provides a picture of the Fe (dobdc) extended unit cell (supercell with 108 atoms) as well as the spin states computed to extract the magnetic coupling values. Equations S3-S6 are the Heisenberg-Dirac-Van Vleck (HDV) Hamiltonian equations for the extended unit cell calculations. The terms of the equations are similar to the ones described in the main paper. In particular this section assumes the full interchain coupling model. Figure S17: Extended unit cell (108 atoms) and spin configurations used to compute the magnetic coupling values of Fe (dobdc), Fe (O) (dobdc), and Fe (OH) (dobdc) at the PBE+U level. This unit cell is double the size of the primitive unit cell (4 atoms) along the c-axis. Blue atoms are iron, red are oxygen, gray are carbon, and white are hydrogen. Red and blue circles indicate the upward or downward spin of the high-spin iron ions. The entirely ferromagnetic (FM), intrachain antiferromagnetic (AFM1), interchain antiferromagnetic (AFM), and nearest-neighbor antiferromagnetic (AFM3) spin states were considered with this unit cell. Equations for computing the values of the 108-atom cell of Fe (dobdc) and the 10-atom cell of Fe (O) (dobdc) are shown below:, H HDV, = E, = [1 NN +1 NNN +1 IC ], H HDV, = E, = [ 1 NN +1 NNN +6 IC ], H HDV, = E, = [ 4 NN 4 NNN +1 IC ], H HDV, = E, = [1 NN +1 NNN 1 IC ] (S11) IC = 1 19 E E,,
19 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-19 NN = 1 19 E, E, 1 4 IC NNN = 1 18 E, E, NN (S1) Equations for computing the values of the doubled cell of Fe (OH) (dobdc) are shown below: ] 1 1 [1,, IC NNN NN, HDV + + = = E H ] [,, IC NNN NN, HDV + + = = E H ] [,, IC NNN NN, HDV + = = E H ] 18 1 [1,, IC NNN NN, HDV + = = E H (S13) [ ],, IC E E = [ ] IC,, NN E E = [ ] NN,, NNN 00 1 E E = (S14)
20 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-0 9. Cluster Calculations Three iron cluster models The coordinates for the cluster models used to compute the nearest-neighbor coupling values are provided in Section S1. The equations used to compute the NN coupling values are provided by eqs S1 and S16. The equations for the Fe (dobdc) and Fe (O) (dobdc) three iron cluster model are, HHDV = E= [ NN ] HHDV = E = [ NN ] NN = 1 [ 3 E E ] (S1) and the equations used for Fe (OH) (dobdc) are, H H NN HDV HDV = 1 0 E = E = [ = E = [ E NN NN ] ] (S16) The equations used to extract both NN and NNN coupling values using the cluster models are as follows: For Fe (dobdc) and Fe (O) (dobdc), H HDV = E = [ NN1 + NN + NNN ] H HDV = E = [ NN1 NN NNN ] H HDV = E = [ NN1 NN + NNN ] H HDV = E = [ NN1 + NN NNN ] (S17) NN1 = 1 3 [ E E + E E ] [ E E ] NN = 16 NN1
21 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S-1 ] [ 1 NN NN1 NN + = [ ] NN NNN E E = (S18) For Fe (OH) (dobdc) they are, ] [ NNN NN NN1 HDV + + = = E H ] [ NNN NN NN1 HDV = = E H ] [ NNN NN NN1 HDV + = = E H ] [ NNN NN NN1 HDV + = = E H (S19) + = NN1 0 1 E E E E + = NN1 NN 16 1 E E ] [ 1 NN NN1 NN + = = NN NNN 16 1 E E (S0) NN1 and NN refer to the two nearest-neighbor interactions in the cluster models. NN is obtained by taking average of NN1 and NN.
22 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S- Two iron cluster models The equations used to compute only NN coupling values are provided by eqs S1 and S. The equations for the Fe (dobdc) two iron cluster model is, NN = 1 16 E E (S1) The equations for the Fe (OH) (dobdc) two iron cluster model is, NN = 1 E E (S)
23 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S Optimized Periodic Structures (VASP Format) Fe (dobdc)-hse O C H Fe Direct
24 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S Fe (dobdc)-hse-d O C H Fe Direct
25 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S Fe (dobdc)-pbe O C H Fe Direct
26 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S Fe (dobdc)-pbe+u
27 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S O C H Fe Direct
28 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S Fe (dobdc)-pbe+u-d O C H Fe Direct
29 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S Fe (O) (dobdc)-hse O O C H Fe Direct
30 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S
31 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S Fe (O) (dobdc)-hse-d O O C H Fe Direct
32 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S Fe (O) (dobdc)-pbe O O C H Fe Direct
33 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S Fe (O) (dobdc)-pbe+u
34 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S O O C H Fe Direct
35 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S Fe (O) (dobdc)-pbe+u-d O O C H Fe Direct
36 Supporting information for paper in Inorganic Chemistry, April 11, 016, page S Fe (OH) (dobdc)-hse O O C H Fe Direct
Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type
 Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type Zhigang Gu, a Lars Heinke, a,* Christof Wöll a, Tobias Neumann,
Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type Zhigang Gu, a Lars Heinke, a,* Christof Wöll a, Tobias Neumann,
Supplementary Information
 Supplementary Information Supplementary Figure S1: Structure and composition of Teflon tape. (a) XRD spectra of original Teflon tape and Teflon tape subjected to annealing at 150 o C under Ar atmosphere.
Supplementary Information Supplementary Figure S1: Structure and composition of Teflon tape. (a) XRD spectra of original Teflon tape and Teflon tape subjected to annealing at 150 o C under Ar atmosphere.
Supporting information. Realizing Two-Dimensional Magnetic Semiconductors with. Enhanced Curie Temperature by Antiaromatic Ring Based
 Supporting information Realizing Two-Dimensional Magnetic Semiconductors with Enhanced Curie Temperature by Antiaromatic Ring Based Organometallic Frameworks Xingxing Li and Jinlong Yang* Department of
Supporting information Realizing Two-Dimensional Magnetic Semiconductors with Enhanced Curie Temperature by Antiaromatic Ring Based Organometallic Frameworks Xingxing Li and Jinlong Yang* Department of
Design of Efficient Catalysts with Double Transition Metal. Atoms on C 2 N Layer
 Supporting Information Design of Efficient Catalysts with Double Transition Metal Atoms on C 2 N Layer Xiyu Li, 1, Wenhui Zhong, 2, Peng Cui, 1 Jun Li, 1 Jun Jiang 1, * 1 Hefei National Laboratory for
Supporting Information Design of Efficient Catalysts with Double Transition Metal Atoms on C 2 N Layer Xiyu Li, 1, Wenhui Zhong, 2, Peng Cui, 1 Jun Li, 1 Jun Jiang 1, * 1 Hefei National Laboratory for
Direct visualization of the Jahn Teller effect coupled to Na ordering in Na 5/8 MnO 2
 Direct visualization of the Jahn Teller effect coupled to Na ordering in Na 5/8 MnO 2 Xin Li 1, Xiaohua Ma 1, Dong Su 2, Lei Liu 1, Robin Chisnell 3, Shyue Ping Ong 1, Hailong Chen 1, Alexandra Toumar
Direct visualization of the Jahn Teller effect coupled to Na ordering in Na 5/8 MnO 2 Xin Li 1, Xiaohua Ma 1, Dong Su 2, Lei Liu 1, Robin Chisnell 3, Shyue Ping Ong 1, Hailong Chen 1, Alexandra Toumar
Defects in TiO 2 Crystals
 , March 13-15, 2013, Hong Kong Defects in TiO 2 Crystals Richard Rivera, Arvids Stashans 1 Abstract-TiO 2 crystals, anatase and rutile, have been studied using Density Functional Theory (DFT) and the Generalized
, March 13-15, 2013, Hong Kong Defects in TiO 2 Crystals Richard Rivera, Arvids Stashans 1 Abstract-TiO 2 crystals, anatase and rutile, have been studied using Density Functional Theory (DFT) and the Generalized
shows the difference between observed (black) and calculated patterns (red). Vertical ticks indicate
 and calculated patterns (red). Vertical ticks indicate") Intensity (arb. unit) a 5 K No disorder Mn-Pt disorder 5 K Mn-Ga disorder 5 K b 5 K Observed Calculated Difference Bragg positions 24 28 32 2 4 6 8 2 4 2θ (degree) 2θ (degree) Supplementary Figure. Powder
Intensity (arb. unit) a 5 K No disorder Mn-Pt disorder 5 K Mn-Ga disorder 5 K b 5 K Observed Calculated Difference Bragg positions 24 28 32 2 4 6 8 2 4 2θ (degree) 2θ (degree) Supplementary Figure. Powder
3.091 Introduction to Solid State Chemistry. Lecture Notes No. 5a ELASTIC BEHAVIOR OF SOLIDS
 3.091 Introduction to Solid State Chemistry Lecture Notes No. 5a ELASTIC BEHAVIOR OF SOLIDS 1. INTRODUCTION Crystals are held together by interatomic or intermolecular bonds. The bonds can be covalent,
3.091 Introduction to Solid State Chemistry Lecture Notes No. 5a ELASTIC BEHAVIOR OF SOLIDS 1. INTRODUCTION Crystals are held together by interatomic or intermolecular bonds. The bonds can be covalent,
Density Functional Theory and the Quantum Chemistry of Gas Separation, Magnetism, and Catalysis in Metal Organic Frameworks
 Density Functional Theory and the Quantum Chemistry of Gas Separation, Magnetism, and Catalysis in Metal Organic Frameworks A DISSERTATION SUBMITTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF MINNESOTA
Density Functional Theory and the Quantum Chemistry of Gas Separation, Magnetism, and Catalysis in Metal Organic Frameworks A DISSERTATION SUBMITTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF MINNESOTA
Magnetic ordering of local moments
 Magnetic ordering Types of magnetic structure Ground state of the Heisenberg ferromagnet and antiferromagnet Spin wave High temperature susceptibility Mean field theory Magnetic ordering of local moments
Magnetic ordering Types of magnetic structure Ground state of the Heisenberg ferromagnet and antiferromagnet Spin wave High temperature susceptibility Mean field theory Magnetic ordering of local moments
Supporting Information
 Supporting Information The Origin of Active Oxygen in a Ternary CuO x /Co 3 O 4 -CeO Catalyst for CO Oxidation Zhigang Liu, *, Zili Wu, *, Xihong Peng, ++ Andrew Binder, Songhai Chai, Sheng Dai *,, School
Supporting Information The Origin of Active Oxygen in a Ternary CuO x /Co 3 O 4 -CeO Catalyst for CO Oxidation Zhigang Liu, *, Zili Wu, *, Xihong Peng, ++ Andrew Binder, Songhai Chai, Sheng Dai *,, School
Observation of a robust zero-energy bound state in iron-based superconductor Fe(Te,Se)
") Materials and Methods: SUPPLEMENTARY INFORMATION Observation of a robust zero-energy bound state in iron-based superconductor Fe(Te,Se) All the crystals, with nominal composition FeTe0.5Se0.5, used in
Materials and Methods: SUPPLEMENTARY INFORMATION Observation of a robust zero-energy bound state in iron-based superconductor Fe(Te,Se) All the crystals, with nominal composition FeTe0.5Se0.5, used in
The electronic structure of materials 1
 Quantum mechanics 2 - Lecture 9 December 18, 2013 1 An overview 2 Literature Contents 1 An overview 2 Literature Electronic ground state Ground state cohesive energy equilibrium crystal structure phase
Quantum mechanics 2 - Lecture 9 December 18, 2013 1 An overview 2 Literature Contents 1 An overview 2 Literature Electronic ground state Ground state cohesive energy equilibrium crystal structure phase
size (nm) Supplementary Figure 1. Hydrodynamic size distribution in a H 2O:MeOH 50% v/v
 Supplementary Figure 1. Hydrodynamic size distribution in a H 2O:MeOH 50% v/v") Intensity (%) 20 15 10 [0a] [0b] CoTPMA: 3 eq CoTPMA: 10 eq CoTPMA: 20 eq CoTPMA : 40 eq CoTPMA: 60 eq, [1] CoTPMA: 85 eq CoTPMA: 125 eq CoTPMA: 170 eq CoTPMA: 210 eq 5 0 10 100 size (nm) Supplementary
Intensity (%) 20 15 10 [0a] [0b] CoTPMA: 3 eq CoTPMA: 10 eq CoTPMA: 20 eq CoTPMA : 40 eq CoTPMA: 60 eq, [1] CoTPMA: 85 eq CoTPMA: 125 eq CoTPMA: 170 eq CoTPMA: 210 eq 5 0 10 100 size (nm) Supplementary
NOT FOR DISTRIBUTION REVIEW COPY. Na 2 IrO 3 as a molecular orbital crystal
 Na 2 IrO 3 as a molecular orbital crystal I. I. Mazin, 1 Harald O. Jeschke, 2 Kateryna Foyevtsova, 2 Roser Valentí, 2 and D. I. Khomskii 3 1 Code 6393, Naval Research Laboratory, Washington, DC 237, USA
Na 2 IrO 3 as a molecular orbital crystal I. I. Mazin, 1 Harald O. Jeschke, 2 Kateryna Foyevtsova, 2 Roser Valentí, 2 and D. I. Khomskii 3 1 Code 6393, Naval Research Laboratory, Washington, DC 237, USA
Supplementary Information for. Universal elastic-hardening-driven mechanical instability in α-quartz and quartz. homeotypes under pressure
 Supplementary Information for Universal elastic-hardening-driven mechanical instability in α-quartz and quartz homeotypes under pressure Juncai Dong, Hailiang Zhu, and Dongliang Chen * Beijing Synchrotron
Supplementary Information for Universal elastic-hardening-driven mechanical instability in α-quartz and quartz homeotypes under pressure Juncai Dong, Hailiang Zhu, and Dongliang Chen * Beijing Synchrotron
Supporting Information. Dissociative Water Adsorption by Al 3 O 4 + in the Gas Phase. Linnéstrasse 2, D Leipzig, Germany.
 Supporting Information Dissociative Water Adsorption by Al 3 O 4 + in the Gas Phase Matias R. Fagiani, 1,2, Xiaowei Song, 1,2 Sreekanta Debnath, 1,2 Sandy Gewinner, 2 Wieland Schöllkopf, 2 Knut R. Asmis,
Supporting Information Dissociative Water Adsorption by Al 3 O 4 + in the Gas Phase Matias R. Fagiani, 1,2, Xiaowei Song, 1,2 Sreekanta Debnath, 1,2 Sandy Gewinner, 2 Wieland Schöllkopf, 2 Knut R. Asmis,
Quantum Chemical Study of Defective Chromium Oxide
 , March 13-15, 2013, Hong Kong Quantum Chemical Study of Defective Chromium Oxide Richard Rivera, Soraya Jácome, Frank Maldonado, Arvids Stashans 1 Abstract Through the use of first-principles calculations
, March 13-15, 2013, Hong Kong Quantum Chemical Study of Defective Chromium Oxide Richard Rivera, Soraya Jácome, Frank Maldonado, Arvids Stashans 1 Abstract Through the use of first-principles calculations
Magnetic properties of spherical fcc clusters with radial surface anisotropy
 Magnetic properties of spherical fcc clusters with radial surface anisotropy D. A. Dimitrov and G. M. Wysin Department of Physics Kansas State University Manhattan, KS 66506-2601 (December 6, 1994) We
Magnetic properties of spherical fcc clusters with radial surface anisotropy D. A. Dimitrov and G. M. Wysin Department of Physics Kansas State University Manhattan, KS 66506-2601 (December 6, 1994) We
Electronic supplementary information (ESI) for. Completing a family: LiCN 3 H 4, the lightest alkali metal guanidinate
 for. Completing a family: LiCN 3 H 4, the lightest alkali metal guanidinate") Electronic supplementary information (ESI) for Completing a family: LiCN 3 H 4, the lightest alkali metal guanidinate Peter Klaus Sawinski, a Volker L. Deringer a and Richard Dronskowski a,b, * a Institute
Electronic supplementary information (ESI) for Completing a family: LiCN 3 H 4, the lightest alkali metal guanidinate Peter Klaus Sawinski, a Volker L. Deringer a and Richard Dronskowski a,b, * a Institute
ELECTRONIC AND MAGNETIC PROPERTIES OF BERKELIUM MONONITRIDE BKN: A FIRST- PRINCIPLES STUDY
 ELECTRONIC AND MAGNETIC PROPERTIES OF BERKELIUM MONONITRIDE BKN: A FIRST- PRINCIPLES STUDY Gitanjali Pagare Department of Physics, Sarojini Naidu Govt. Girls P. G. Auto. College, Bhopal ( India) ABSTRACT
ELECTRONIC AND MAGNETIC PROPERTIES OF BERKELIUM MONONITRIDE BKN: A FIRST- PRINCIPLES STUDY Gitanjali Pagare Department of Physics, Sarojini Naidu Govt. Girls P. G. Auto. College, Bhopal ( India) ABSTRACT
Minnesota Functional Module Version 1.8
 1 Minnesota Functional Module Version 1.8 Subroutines for evaluating the M05, M05-2X, M06-L, M06-HF, M06, M06-2X, M08-HX, M08-SO, M11, M11-L, MN12-L, SOGGA, SOGGA11, SOGGA11-X, N12, N12-SX Functionals
1 Minnesota Functional Module Version 1.8 Subroutines for evaluating the M05, M05-2X, M06-L, M06-HF, M06, M06-2X, M08-HX, M08-SO, M11, M11-L, MN12-L, SOGGA, SOGGA11, SOGGA11-X, N12, N12-SX Functionals
2.1 Experimental and theoretical studies
 Chapter 2 NiO As stated before, the first-row transition-metal oxides are among the most interesting series of materials, exhibiting wide variations in physical properties related to electronic structure.
Chapter 2 NiO As stated before, the first-row transition-metal oxides are among the most interesting series of materials, exhibiting wide variations in physical properties related to electronic structure.
Minnesota Functional Module Version 2.0
 1 Minnesota Functional Module Version 2.0 Subroutines for evaluating the M05, M05-2X, M06-L, M06-HF, M06, M06-2X, M08-HX, M08-SO, M11, M11-L, MN12-L, GAM, MN15-L, MN15, SOGGA, SOGGA11, SOGGA11-X, N12,
1 Minnesota Functional Module Version 2.0 Subroutines for evaluating the M05, M05-2X, M06-L, M06-HF, M06, M06-2X, M08-HX, M08-SO, M11, M11-L, MN12-L, GAM, MN15-L, MN15, SOGGA, SOGGA11, SOGGA11-X, N12,
SUPPLEMENTARY INFORMATION
 Atomic structure and dynamic behaviour of truly one-dimensional ionic chains inside carbon nanotubes Ryosuke Senga 1, Hannu-Pekka Komsa 2, Zheng Liu 1, Kaori Hirose-Takai 1, Arkady V. Krasheninnikov 2
Atomic structure and dynamic behaviour of truly one-dimensional ionic chains inside carbon nanotubes Ryosuke Senga 1, Hannu-Pekka Komsa 2, Zheng Liu 1, Kaori Hirose-Takai 1, Arkady V. Krasheninnikov 2
Magnetism in transition metal oxides by post-dft methods
 Magnetism in transition metal oxides by post-dft methods Cesare Franchini Faculty of Physics & Center for Computational Materials Science University of Vienna, Austria Workshop on Magnetism in Complex
Magnetism in transition metal oxides by post-dft methods Cesare Franchini Faculty of Physics & Center for Computational Materials Science University of Vienna, Austria Workshop on Magnetism in Complex
Single-Ion Magnetic Anisotropy and Isotropic Magnetic Couplings in the Metal Organic Framework Fe 2 (dobdc)
") pubs.acs.org/ic Single-Ion Magnetic Anisotropy and Isotropic Magnetic Couplings in the Metal Organic Framework Fe (dobdc) Reḿi Maurice, Pragya Verma, Joseph M. Zadrozny, Sijie Luo, Joshua Borycz, Jeffrey
pubs.acs.org/ic Single-Ion Magnetic Anisotropy and Isotropic Magnetic Couplings in the Metal Organic Framework Fe (dobdc) Reḿi Maurice, Pragya Verma, Joseph M. Zadrozny, Sijie Luo, Joshua Borycz, Jeffrey
Multiscale Model for a Metal-Organic Framework: High-Spin Rebound Mechanism in the Reaction of the Oxoiron(IV) Species of Fe-MOF-74 Contents
 Species of Fe-MOF-74 Contents") Supporting Information Multiscale Model for a Metal-Organic Framework: High-Spin Rebound Mechanism in the Reaction of the Oxoiron(IV) Species of Fe-MOF-74 Hajime Hirao*, Wilson Kwok Hung Ng, Adhitya Mangala
Supporting Information Multiscale Model for a Metal-Organic Framework: High-Spin Rebound Mechanism in the Reaction of the Oxoiron(IV) Species of Fe-MOF-74 Hajime Hirao*, Wilson Kwok Hung Ng, Adhitya Mangala
Energy-Level Alignment at the Interface of Graphene Fluoride and Boron Nitride Monolayers: An Investigation by Many-Body Perturbation Theory
 Supporting Information Energy-Level Alignment at the Interface of Graphene Fluoride and Boron Nitride Monolayers: An Investigation by Many-Body Perturbation Theory Qiang Fu, Dmitrii Nabok, and Claudia
Supporting Information Energy-Level Alignment at the Interface of Graphene Fluoride and Boron Nitride Monolayers: An Investigation by Many-Body Perturbation Theory Qiang Fu, Dmitrii Nabok, and Claudia
SnO 2 Physical and Chemical Properties due to the Impurity Doping
 , March 13-15, 2013, Hong Kong SnO 2 Physical and Chemical Properties due to the Impurity Doping Richard Rivera, Freddy Marcillo, Washington Chamba, Patricio Puchaicela, Arvids Stashans Abstract First-principles
, March 13-15, 2013, Hong Kong SnO 2 Physical and Chemical Properties due to the Impurity Doping Richard Rivera, Freddy Marcillo, Washington Chamba, Patricio Puchaicela, Arvids Stashans Abstract First-principles
Helicoidal magnetic structure and ferroelectric polarization in. Cu 3 Nb 2 O 8
 Helicoidal magnetic structure and ferroelectric polarization in Cu 3 Nb 2 O 8 Zheng-Lu Li, 1 M.-H. Whangbo, 2 X. G. Gong, 1, * and H. J. Xiang 1, * 1 Key Laboratory of Computational Physical Sciences (Ministry
Helicoidal magnetic structure and ferroelectric polarization in Cu 3 Nb 2 O 8 Zheng-Lu Li, 1 M.-H. Whangbo, 2 X. G. Gong, 1, * and H. J. Xiang 1, * 1 Key Laboratory of Computational Physical Sciences (Ministry
EFFECTIVE MAGNETIC HAMILTONIANS: ab initio determination
 ICSM212, Istanbul, May 3, 212, Theoretical Magnetism I, 17:2 p. 1 EFFECTIVE MAGNETIC HAMILTONIANS: ab initio determination Václav Drchal Institute of Physics ASCR, Praha, Czech Republic in collaboration
ICSM212, Istanbul, May 3, 212, Theoretical Magnetism I, 17:2 p. 1 EFFECTIVE MAGNETIC HAMILTONIANS: ab initio determination Václav Drchal Institute of Physics ASCR, Praha, Czech Republic in collaboration
Hybrid density functional theory applied to magnetite: Crystal structure, charge order, and phonons
 Hybrid density functional theory applied to magnetite: Crystal structure, charge order, and phonons Andrew D. Rowan and Charles H. Patterson School of Physics, Trinity College Dublin, Dublin 2, Ireland
Hybrid density functional theory applied to magnetite: Crystal structure, charge order, and phonons Andrew D. Rowan and Charles H. Patterson School of Physics, Trinity College Dublin, Dublin 2, Ireland
Observation of quadrupole helix chirality and its domain structure in DyFe 3 (BO 3 ) 4
 4") Observation of quadrupole helix chirality and its domain structure in DyFe 3 (BO 3 ) 4 T. Usui, Y. Tanaka, H. Nakajima, M. Taguchi, A. Chainani, M. Oura, S. Shin, N. Katayama, H. Sawa, Y. Wakabayashi,
Observation of quadrupole helix chirality and its domain structure in DyFe 3 (BO 3 ) 4 T. Usui, Y. Tanaka, H. Nakajima, M. Taguchi, A. Chainani, M. Oura, S. Shin, N. Katayama, H. Sawa, Y. Wakabayashi,
ELECTRONIC SUPPLEMENTARY INFORMATION
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2017 S-1 ELECTRONIC SUPPLEMENTARY INFORMATION OCT. 1, 2017 Combined Quantum Mechanical
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2017 S-1 ELECTRONIC SUPPLEMENTARY INFORMATION OCT. 1, 2017 Combined Quantum Mechanical
SUPPLEMENTARY INFORMATION
 Supplementary Methods Materials Synthesis The In 4 Se 3-δ crystal ingots were grown by the Bridgeman method. The In and Se elements were placed in an evacuated quartz ampoule with an excess of In (5-10
Supplementary Methods Materials Synthesis The In 4 Se 3-δ crystal ingots were grown by the Bridgeman method. The In and Se elements were placed in an evacuated quartz ampoule with an excess of In (5-10
Stable Ferromagnetism and Half-metallicity in Two-Dimensional. Polyporphyrin Frameworks
 Supporting Information for Stable Ferromagnetism and Half-metallicity in Two-Dimensional Polyporphyrin Frameworks Jie Tan, a Weifeng Li, b Xiujie He, a Mingwen Zhao a,* a School of Physics and State Key
Supporting Information for Stable Ferromagnetism and Half-metallicity in Two-Dimensional Polyporphyrin Frameworks Jie Tan, a Weifeng Li, b Xiujie He, a Mingwen Zhao a,* a School of Physics and State Key
Ab initio evaluation of Coulomb and exchange parameters for DFT+ U calculations
 PHYSICAL REVIEW B 7, 15513 7 Ab initio evaluation of Coulomb and exchange parameters for DFT+ U calculations Nicholas J. Mosey and Emily A. Carter* Department of Mechanical and Aerospace Engineering and
PHYSICAL REVIEW B 7, 15513 7 Ab initio evaluation of Coulomb and exchange parameters for DFT+ U calculations Nicholas J. Mosey and Emily A. Carter* Department of Mechanical and Aerospace Engineering and
SUPPLEMENTARY INFORMATION
 DOI: 10.1038/NCHEM.1896 Unusual Structure, Bonding and Properties in a Californium Borate Matthew J. Polinski, 1 Edward B. Garner, III, 2 Rémi Maurice, 3,4 Nora Planas, 3 Jared T. Stritzinger, 1 T. Gannon
DOI: 10.1038/NCHEM.1896 Unusual Structure, Bonding and Properties in a Californium Borate Matthew J. Polinski, 1 Edward B. Garner, III, 2 Rémi Maurice, 3,4 Nora Planas, 3 Jared T. Stritzinger, 1 T. Gannon
Outline. Introduction: graphene. Adsorption on graphene: - Chemisorption - Physisorption. Summary
 Outline Introduction: graphene Adsorption on graphene: - Chemisorption - Physisorption Summary 1 Electronic band structure: Electronic properties K Γ M v F = 10 6 ms -1 = c/300 massless Dirac particles!
Outline Introduction: graphene Adsorption on graphene: - Chemisorption - Physisorption Summary 1 Electronic band structure: Electronic properties K Γ M v F = 10 6 ms -1 = c/300 massless Dirac particles!
Crystal Relaxation, Elasticity, and Lattice Dynamics
 http://exciting-code.org Crystal Relaxation, Elasticity, and Lattice Dynamics Pasquale Pavone Humboldt-Universität zu Berlin http://exciting-code.org PART I: Structure Optimization Pasquale Pavone Humboldt-Universität
http://exciting-code.org Crystal Relaxation, Elasticity, and Lattice Dynamics Pasquale Pavone Humboldt-Universität zu Berlin http://exciting-code.org PART I: Structure Optimization Pasquale Pavone Humboldt-Universität
Structure, energetics, and mechanical stability of Fe-Cu bcc alloys from first-principles calculations
 Structure, energetics, and mechanical stability of Fe-Cu bcc alloys from first-principles calculations Jefferson Z. Liu, A. van de Walle, G. Ghosh, and M. Asta* Department of Materials Science and Engineering,
Structure, energetics, and mechanical stability of Fe-Cu bcc alloys from first-principles calculations Jefferson Z. Liu, A. van de Walle, G. Ghosh, and M. Asta* Department of Materials Science and Engineering,
Computational support for a pyrolitic lower mantle containing ferric iron
 SUPPLEMENTARY INFORMATION DOI: 1.138/NGEO2458 Computational support for a pyrolitic lower mantle containing ferric iron Xianlong Wang, Taku Tsuchiya and Atsushi Hase NATURE GEOSCIENCE www.nature.com/naturegeoscience
SUPPLEMENTARY INFORMATION DOI: 1.138/NGEO2458 Computational support for a pyrolitic lower mantle containing ferric iron Xianlong Wang, Taku Tsuchiya and Atsushi Hase NATURE GEOSCIENCE www.nature.com/naturegeoscience
Unconventional magnetic order in 3D Kitaev materials revealed by resonant x-ray diffraction Radu Coldea
 Unconventional magnetic order in 3D Kitaev materials revealed by resonant x-ray diffraction Radu Coldea Oxford Collaborators Alun Biffin (Oxford->PSI) Roger D. Johnson S. Choi P. Manuel A. Bombardi Sample
Unconventional magnetic order in 3D Kitaev materials revealed by resonant x-ray diffraction Radu Coldea Oxford Collaborators Alun Biffin (Oxford->PSI) Roger D. Johnson S. Choi P. Manuel A. Bombardi Sample
International Journal of Quantum Chemistry
 International Journal of Quantum Chemistry First-principles calculation of second-order elastic constants and equations of state for Lithium Azide, LiN, and Lead Azide, Pb(N ) Journal: International Journal
International Journal of Quantum Chemistry First-principles calculation of second-order elastic constants and equations of state for Lithium Azide, LiN, and Lead Azide, Pb(N ) Journal: International Journal
Materials Chemistry A
 Journal of Materials Chemistry A PAPER Cite this: J. Mater. Chem. A, 2015,3, 8002 Received 22nd December 2014 Accepted 4th March 2015 DOI: 10.1039/c4ta07062c www.rsc.org/materialsa View Journal View Issue
Journal of Materials Chemistry A PAPER Cite this: J. Mater. Chem. A, 2015,3, 8002 Received 22nd December 2014 Accepted 4th March 2015 DOI: 10.1039/c4ta07062c www.rsc.org/materialsa View Journal View Issue
Supporting Information
 Electronic Supplementary Material (ESI) for Journal of Materials Chemistry A. This journal is The Royal Society of Chemistry 2018 Supporting Information Tuning thermoelectric performance of π-d conjugated
Electronic Supplementary Material (ESI) for Journal of Materials Chemistry A. This journal is The Royal Society of Chemistry 2018 Supporting Information Tuning thermoelectric performance of π-d conjugated
Hydrogen-bonded structure and mechanical chiral response of a silver nanoparticle superlattice
 Hydrogen-bonded structure and mechanical chiral response of a silver nanoparticle superlattice Bokwon Yoon 1, W. D. Luedtke 1, Robert N. Barnett 1, Jianping Gao 1, Anil Desireddy 2, Brian E. Conn 2, Terry
Hydrogen-bonded structure and mechanical chiral response of a silver nanoparticle superlattice Bokwon Yoon 1, W. D. Luedtke 1, Robert N. Barnett 1, Jianping Gao 1, Anil Desireddy 2, Brian E. Conn 2, Terry
Supporting information for Polymer interactions with Reduced Graphene Oxide: Van der Waals binding energies of Benzene on defected Graphene
 Supporting information for Polymer interactions with Reduced Graphene Oxide: Van der Waals binding energies of Benzene on defected Graphene Mohamed Hassan, Michael Walter *,,, and Michael Moseler, Freiburg
Supporting information for Polymer interactions with Reduced Graphene Oxide: Van der Waals binding energies of Benzene on defected Graphene Mohamed Hassan, Michael Walter *,,, and Michael Moseler, Freiburg
Supplementary Information
 Supplementary Information Supplementary Figure 1: After structural optimization of the CH 3 NH 3 PbI 3 unit cell, the resulting relaxed volumes for three distinct orientation of the MA molecules are shown.
Supplementary Information Supplementary Figure 1: After structural optimization of the CH 3 NH 3 PbI 3 unit cell, the resulting relaxed volumes for three distinct orientation of the MA molecules are shown.
Single-Layer Tl 2 O: A Metal-Shrouded 2D Semiconductor with High Electronic Mobility
 Supporting information for Single-Layer Tl 2 O: A Metal-Shrouded 2D Semiconductor with High Electronic Mobility Yandong Ma, Agnieszka Kuc, and Thomas Heine*, Wilhelm-Ostwald-Institut für Physikalische
Supporting information for Single-Layer Tl 2 O: A Metal-Shrouded 2D Semiconductor with High Electronic Mobility Yandong Ma, Agnieszka Kuc, and Thomas Heine*, Wilhelm-Ostwald-Institut für Physikalische
Figure 1. Oxidation by iron-oxo complex. supported by porous solid
 Oxidation of Ethane to Ethanol by N 2 O in a Metal-Organic Framework with Coordinatively Unsaturated Iron(II) Sites Long, J.R, et al., Nat. Chem. 2014, 6, 590. Mechanism of Oxidation of Ethane to Ethanol
Oxidation of Ethane to Ethanol by N 2 O in a Metal-Organic Framework with Coordinatively Unsaturated Iron(II) Sites Long, J.R, et al., Nat. Chem. 2014, 6, 590. Mechanism of Oxidation of Ethane to Ethanol
Supporting Information
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2015 Supporting Information Single Layer Lead Iodide: Computational Exploration of Structural, Electronic
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2015 Supporting Information Single Layer Lead Iodide: Computational Exploration of Structural, Electronic
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
Tinselenidene: a Two-dimensional Auxetic Material with Ultralow Lattice Thermal Conductivity and Ultrahigh Hole Mobility
 Tinselenidene: a Two-dimensional Auxetic Material with Ultralow Lattice Thermal Conductivity and Ultrahigh Hole Mobility Li-Chuan Zhang, Guangzhao Qin, Wu-Zhang Fang, Hui-Juan Cui, Qing-Rong Zheng, Qing-Bo
Tinselenidene: a Two-dimensional Auxetic Material with Ultralow Lattice Thermal Conductivity and Ultrahigh Hole Mobility Li-Chuan Zhang, Guangzhao Qin, Wu-Zhang Fang, Hui-Juan Cui, Qing-Rong Zheng, Qing-Bo
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory
 Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
DFT EXERCISES. FELIPE CERVANTES SODI January 2006
 DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
arxiv: v1 [cond-mat.mtrl-sci] 21 Dec 2007
![arxiv: v1 [cond-mat.mtrl-sci] 21 Dec 2007](/thumbs/92/110189214.jpg "arxiv: v1 [cond-mat.mtrl-sci] 21 Dec 2007") Quantum Monte Carlo calculations of structural properties of FeO under pressure Jindřich Kolorenč and Lubos Mitas Department of Physics and CHiPS, North Carolina State University, Raleigh, North Carolina
Quantum Monte Carlo calculations of structural properties of FeO under pressure Jindřich Kolorenč and Lubos Mitas Department of Physics and CHiPS, North Carolina State University, Raleigh, North Carolina
MO Calculation for a Diatomic Molecule. /4 0 ) i=1 j>i (1/r ij )
 i=1 j>i (1/r ij )") MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
First-Principles Calculation of Exchange Interactions
 Chapter 2 First-Principles Calculation of Exchange Interactions Before introducing the first-principles methods for the calculation of exchange interactions in magnetic systems we will briefly review two
Chapter 2 First-Principles Calculation of Exchange Interactions Before introducing the first-principles methods for the calculation of exchange interactions in magnetic systems we will briefly review two
Crystalline and Magnetic Anisotropy of the 3d Transition-Metal Monoxides
 Crystalline and of the 3d Transition-Metal Monoxides Institut für Festkörpertheorie und -optik Friedrich-Schiller-Universität Max-Wien-Platz 1 07743 Jena 2012-03-23 Introduction Crystalline Anisotropy
Crystalline and of the 3d Transition-Metal Monoxides Institut für Festkörpertheorie und -optik Friedrich-Schiller-Universität Max-Wien-Platz 1 07743 Jena 2012-03-23 Introduction Crystalline Anisotropy
Reactivity of Diiron Imido Complexes
 One-electron Oxidation Chemistry and Subsequent Reactivity of Diiron Imido Complexes Subramaniam Kuppuswamy, Tamara M. Powers, Bruce M. Johnson, Carl K. Brozek, Jeremy P. Krogman, Mark W. Bezpalko, Louise
One-electron Oxidation Chemistry and Subsequent Reactivity of Diiron Imido Complexes Subramaniam Kuppuswamy, Tamara M. Powers, Bruce M. Johnson, Carl K. Brozek, Jeremy P. Krogman, Mark W. Bezpalko, Louise
Introduction to Solid State Physics or the study of physical properties of matter in a solid phase
 Introduction to Solid State Physics or the study of physical properties of matter in a solid phase Prof. Germar Hoffmann 1. Crystal Structures 2. Reciprocal Lattice 3. Crystal Binding and Elastic Constants
Introduction to Solid State Physics or the study of physical properties of matter in a solid phase Prof. Germar Hoffmann 1. Crystal Structures 2. Reciprocal Lattice 3. Crystal Binding and Elastic Constants
The Oxford Solid State Basics
 The Oxford Solid State Basics Steven H. Simon University of Oxford OXFORD UNIVERSITY PRESS Contents 1 About Condensed Matter Physics 1 1.1 What Is Condensed Matter Physics 1 1.2 Why Do We Study Condensed
The Oxford Solid State Basics Steven H. Simon University of Oxford OXFORD UNIVERSITY PRESS Contents 1 About Condensed Matter Physics 1 1.1 What Is Condensed Matter Physics 1 1.2 Why Do We Study Condensed
Final Exam, MENA3000 / MENA4000 Functional Materials, 6 th June 2016
 Final Exam, MENA3000 / MENA4000 Functional Materials, 6 th June 2016 Task 1 Crystal structure, chemical bonding and non-stoichiometry (25 %) For one of the modifications of molybdenum disulphide, MoS 2,
Final Exam, MENA3000 / MENA4000 Functional Materials, 6 th June 2016 Task 1 Crystal structure, chemical bonding and non-stoichiometry (25 %) For one of the modifications of molybdenum disulphide, MoS 2,
Magnetic Oxides. Gerald F. Dionne. Department of Materials Science and Engineering Massachusetts Institute of Technology
 Magnetic Oxides Gerald F. Dionne Department of Materials Science and Engineering Massachusetts Institute of Technology Spins in Solids Summer School University of Virginia Charlottesville, VA 21 June 2006
Magnetic Oxides Gerald F. Dionne Department of Materials Science and Engineering Massachusetts Institute of Technology Spins in Solids Summer School University of Virginia Charlottesville, VA 21 June 2006
MANUAL Minnesota Functional Module
 1 MANUAL Minnesota Functional Module Version 4.0 Subroutines for evaluating the following exchange-correlation functionals: GAM, M05, M05-2X, M06, M06-2X, M06-HF, M06-L, M08-HX, M08-SO, M11, M11-L, MN12-L,
1 MANUAL Minnesota Functional Module Version 4.0 Subroutines for evaluating the following exchange-correlation functionals: GAM, M05, M05-2X, M06, M06-2X, M06-HF, M06-L, M08-HX, M08-SO, M11, M11-L, MN12-L,
First Principles Calculation of Defect and Magnetic Structures in FeCo
 Materials Transactions, Vol. 47, No. 11 (26) pp. 2646 to 26 Special Issue on Advances in Computational Materials Science and Engineering IV #26 The Japan Institute of Metals First Principles Calculation
Materials Transactions, Vol. 47, No. 11 (26) pp. 2646 to 26 Special Issue on Advances in Computational Materials Science and Engineering IV #26 The Japan Institute of Metals First Principles Calculation
WORLD SCIENTIFIC (2014)
") WORLD SCIENTIFIC (2014) LIST OF PROBLEMS Chapter 1: Magnetism of Free Electrons and Atoms 1. Orbital and spin moments of an electron: Using the theory of angular momentum, calculate the orbital
WORLD SCIENTIFIC (2014) LIST OF PROBLEMS Chapter 1: Magnetism of Free Electrons and Atoms 1. Orbital and spin moments of an electron: Using the theory of angular momentum, calculate the orbital
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić
 Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
PBS: FROM SOLIDS TO CLUSTERS
 PBS: FROM SOLIDS TO CLUSTERS E. HOFFMANN AND P. ENTEL Theoretische Tieftemperaturphysik Gerhard-Mercator-Universität Duisburg, Lotharstraße 1 47048 Duisburg, Germany Semiconducting nanocrystallites like
PBS: FROM SOLIDS TO CLUSTERS E. HOFFMANN AND P. ENTEL Theoretische Tieftemperaturphysik Gerhard-Mercator-Universität Duisburg, Lotharstraße 1 47048 Duisburg, Germany Semiconducting nanocrystallites like
Quantum order-by-disorder in Kitaev model on a triangular lattice
 Quantum order-by-disorder in Kitaev model on a triangular lattice George Jackeli Max-Planck Institute & University of Stuttgart, Germany Andronikashvili Institute of Physics, Tbilisi, Georgia GJ & Avella,
Quantum order-by-disorder in Kitaev model on a triangular lattice George Jackeli Max-Planck Institute & University of Stuttgart, Germany Andronikashvili Institute of Physics, Tbilisi, Georgia GJ & Avella,
Lecture 11: Transition metals (1) Basics and magnetism
 Basics and magnetism") Lecture 11: Transition metals (1) Basics and magnetism Oxidation states in transition metal compounds Ligand field theory Magnetism Susceptibility Temperature dependence Magnetic moments Figure: Wikipedia
Lecture 11: Transition metals (1) Basics and magnetism Oxidation states in transition metal compounds Ligand field theory Magnetism Susceptibility Temperature dependence Magnetic moments Figure: Wikipedia
arxiv: v2 [cond-mat.str-el] 28 Nov 2009
![arxiv: v2 [cond-mat.str-el] 28 Nov 2009](/thumbs/88/117929903.jpg "arxiv: v2 [cond-mat.str-el] 28 Nov 2009") First-principles study of α-pu 2 O 3 Hongliang Shi 1,2 and Ping Zhang 1,3, 1 Institute of Applied Physics and Computational Mathematics, P.O. Box 8009, Beijing 100088, People s Republic of China 2 SKLSM,
First-principles study of α-pu 2 O 3 Hongliang Shi 1,2 and Ping Zhang 1,3, 1 Institute of Applied Physics and Computational Mathematics, P.O. Box 8009, Beijing 100088, People s Republic of China 2 SKLSM,
Structure and Dynamics : An Atomic View of Materials
 Structure and Dynamics : An Atomic View of Materials MARTIN T. DOVE Department ofearth Sciences University of Cambridge OXFORD UNIVERSITY PRESS Contents 1 Introduction 1 1.1 Observations 1 1.1.1 Microscopic
Structure and Dynamics : An Atomic View of Materials MARTIN T. DOVE Department ofearth Sciences University of Cambridge OXFORD UNIVERSITY PRESS Contents 1 Introduction 1 1.1 Observations 1 1.1.1 Microscopic
Chemical bonding, elasticity, and valence force field models: A case study for -Pt 2 Si and PtSi
 PHYSICAL REVIEW B, VOLUME 64, 550 Chemical bonding, elasticity, and valence force field models: A case study for -Pt 2 Si and PtSi J. E. Klepeis, O. Beckstein, 2 O. Pankratov, 2 and G. L. W. Hart 3 Lawrence
PHYSICAL REVIEW B, VOLUME 64, 550 Chemical bonding, elasticity, and valence force field models: A case study for -Pt 2 Si and PtSi J. E. Klepeis, O. Beckstein, 2 O. Pankratov, 2 and G. L. W. Hart 3 Lawrence
Structural Calculations phase stability, surfaces, interfaces etc
 Structural Calculations phase stability, surfaces, interfaces etc Keith Refson STFC Rutherford Appleton Laboratory September 19, 2007 Phase Equilibrium 2 Energy-Volume curves..................................................................
Structural Calculations phase stability, surfaces, interfaces etc Keith Refson STFC Rutherford Appleton Laboratory September 19, 2007 Phase Equilibrium 2 Energy-Volume curves..................................................................
Theoretical study of electronic and atomic structures of (MnO)n
n") Theoretical study of electronic and atomic structures of (MnO)n Hiori Kino, a Lucas K. Wagner b and Lubos Mitas c a National Institute for Materials Science, 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan.
Theoretical study of electronic and atomic structures of (MnO)n Hiori Kino, a Lucas K. Wagner b and Lubos Mitas c a National Institute for Materials Science, 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan.
Supporting Information. Directing the Breathing Behavior of Pillared-Layered. Metal Organic Frameworks via a Systematic Library of
 Supporting Information Directing the Breathing Behavior of Pillared-Layered Metal Organic Frameworks via a Systematic Library of Functionalized Linkers Bearing Flexible Substituents Sebastian Henke, Andreas
Supporting Information Directing the Breathing Behavior of Pillared-Layered Metal Organic Frameworks via a Systematic Library of Functionalized Linkers Bearing Flexible Substituents Sebastian Henke, Andreas
Theoretical and experimental study of aromatic hydrocarbon superconductors
 Workshop on correlations, criticality, and coherence in quantum systems Theoretical and experimental study of aromatic hydrocarbon superconductors Zhongbing Huang 1 Hubei University, Wuhan, China 2 Beijing
Workshop on correlations, criticality, and coherence in quantum systems Theoretical and experimental study of aromatic hydrocarbon superconductors Zhongbing Huang 1 Hubei University, Wuhan, China 2 Beijing
Coupling of Functional Hydrogen Bonds in Pyridoxal-5 -phosphate- Enzyme Model Systems Observed by Solid State NMR
 Supporting Information for Coupling of Functional ydrogen Bonds in Pyridoxal-5 -phosphate- Enzyme Model Systems bserved by Solid State NMR Shasad Sharif, David Schagen, Michael D. Toney, and ans-einrich
Supporting Information for Coupling of Functional ydrogen Bonds in Pyridoxal-5 -phosphate- Enzyme Model Systems bserved by Solid State NMR Shasad Sharif, David Schagen, Michael D. Toney, and ans-einrich
The Quantum Theory of Magnetism
 The Quantum Theory of Magnetism Norberto Mains McGill University, Canada I: 0 World Scientific Singapore NewJersey London Hong Kong Contents 1 Paramagnetism 1.1 Introduction 1.2 Quantum mechanics of atoms
The Quantum Theory of Magnetism Norberto Mains McGill University, Canada I: 0 World Scientific Singapore NewJersey London Hong Kong Contents 1 Paramagnetism 1.1 Introduction 1.2 Quantum mechanics of atoms
Supplementary Figures
 Supplementary Figures 8 6 Energy (ev 4 2 2 4 Γ M K Γ Supplementary Figure : Energy bands of antimonene along a high-symmetry path in the Brillouin zone, including spin-orbit coupling effects. Empty circles
Supplementary Figures 8 6 Energy (ev 4 2 2 4 Γ M K Γ Supplementary Figure : Energy bands of antimonene along a high-symmetry path in the Brillouin zone, including spin-orbit coupling effects. Empty circles
Supporting Information. Don-Hyung Ha, Liane M. Moreau, Clive R. Bealing, Haitao Zhang, Richard G. Hennig, and. Richard D.
 Supporting Information The structural evolution and diffusion during the chemical transformation from cobalt to cobalt phosphide nanoparticles Don-Hyung Ha, Liane M. Moreau, Clive R. Bealing, Haitao Zhang,
Supporting Information The structural evolution and diffusion during the chemical transformation from cobalt to cobalt phosphide nanoparticles Don-Hyung Ha, Liane M. Moreau, Clive R. Bealing, Haitao Zhang,
复习题. 2 Calculate the intensity of magnetic field in the air gap of the magnetic circuit shown in the figure. Use the values N=200,
 复习题 1 Calculate the magnetic moment of a sphere of radius R made from a magnetic material with magnetic susceptibility, when it is magnetized by an external magnetic field H. How is the value of the moment
复习题 1 Calculate the magnetic moment of a sphere of radius R made from a magnetic material with magnetic susceptibility, when it is magnetized by an external magnetic field H. How is the value of the moment
SUPPLEMENTARY INFORMATION
 DOI: 10.1038/NCHEM.1677 Entangled quantum electronic wavefunctions of the Mn 4 CaO 5 cluster in photosystem II Yuki Kurashige 1 *, Garnet Kin-Lic Chan 2, Takeshi Yanai 1 1 Department of Theoretical and
DOI: 10.1038/NCHEM.1677 Entangled quantum electronic wavefunctions of the Mn 4 CaO 5 cluster in photosystem II Yuki Kurashige 1 *, Garnet Kin-Lic Chan 2, Takeshi Yanai 1 1 Department of Theoretical and
SOLID STATE PHYSICS. Second Edition. John Wiley & Sons. J. R. Hook H. E. Hall. Department of Physics, University of Manchester
 SOLID STATE PHYSICS Second Edition J. R. Hook H. E. Hall Department of Physics, University of Manchester John Wiley & Sons CHICHESTER NEW YORK BRISBANE TORONTO SINGAPORE Contents Flow diagram Inside front
SOLID STATE PHYSICS Second Edition J. R. Hook H. E. Hall Department of Physics, University of Manchester John Wiley & Sons CHICHESTER NEW YORK BRISBANE TORONTO SINGAPORE Contents Flow diagram Inside front
Topological edge states in a high-temperature superconductor FeSe/SrTiO 3 (001) film
 film") Topological edge states in a high-temperature superconductor FeSe/SrTiO 3 (001) film Z. F. Wang 1,2,3+, Huimin Zhang 2,4+, Defa Liu 5, Chong Liu 2, Chenjia Tang 2, Canli Song 2, Yong Zhong 2, Junping Peng
Topological edge states in a high-temperature superconductor FeSe/SrTiO 3 (001) film Z. F. Wang 1,2,3+, Huimin Zhang 2,4+, Defa Liu 5, Chong Liu 2, Chenjia Tang 2, Canli Song 2, Yong Zhong 2, Junping Peng
Selectivity in the initial C-H bond cleavage of n-butane on PdO(101)
") Supporting Information for Selectivity in the initial C-H bond cleavage of n-butane on PdO(101) Can Hakanoglu (a), Feng Zhang (a), Abbin Antony (a), Aravind Asthagiri (b) and Jason F. Weaver (a) * (a)
Supporting Information for Selectivity in the initial C-H bond cleavage of n-butane on PdO(101) Can Hakanoglu (a), Feng Zhang (a), Abbin Antony (a), Aravind Asthagiri (b) and Jason F. Weaver (a) * (a)
Hydrogenation of Penta-Graphene Leads to Unexpected Large. Improvement in Thermal Conductivity
 Supplementary information for Hydrogenation of Penta-Graphene Leads to Unexpected Large Improvement in Thermal Conductivity Xufei Wu, a Vikas Varshney, b,c Jonghoon Lee, b,c Teng Zhang, a Jennifer L. Wohlwend,
Supplementary information for Hydrogenation of Penta-Graphene Leads to Unexpected Large Improvement in Thermal Conductivity Xufei Wu, a Vikas Varshney, b,c Jonghoon Lee, b,c Teng Zhang, a Jennifer L. Wohlwend,
SUPPLEMENTARY INFORMATION
 Materials and Methods Single crystals of Pr 2 Ir 2 O 7 were grown by a flux method [S1]. Energy dispersive x-ray analysis found no impurity phases, no inhomogeneities and a ratio between Pr and Ir of 1:1.03(3).
Materials and Methods Single crystals of Pr 2 Ir 2 O 7 were grown by a flux method [S1]. Energy dispersive x-ray analysis found no impurity phases, no inhomogeneities and a ratio between Pr and Ir of 1:1.03(3).
Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data January 2012
 2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
Periodic DFT Study of Molecular Crystals
 , March 13-15, 2013, Hong Kong Periodic DFT Study of Molecular Crystals Richard Rivera, Soraya Jácome, Darwin Castillo, Arvids Stashans 1 Abstract Two molecular crystals have been studied using the first-principles
, March 13-15, 2013, Hong Kong Periodic DFT Study of Molecular Crystals Richard Rivera, Soraya Jácome, Darwin Castillo, Arvids Stashans 1 Abstract Two molecular crystals have been studied using the first-principles
Magnetism of close packed Fe 147 clusters
 Phase Transitions Vol. 00, No. 00, June 2005, 1 10 Magnetism of close packed Fe 147 clusters M. E. Gruner, G. Rollmann, S. Sahoo and P. Entel a Theoretical Physics, Universität Duisburg-Essen, Campus Duisburg,
Phase Transitions Vol. 00, No. 00, June 2005, 1 10 Magnetism of close packed Fe 147 clusters M. E. Gruner, G. Rollmann, S. Sahoo and P. Entel a Theoretical Physics, Universität Duisburg-Essen, Campus Duisburg,
Exchange-induced negative-u charge order in N-doped WO 3 : A spin-peierls-like system
 Exchange-induced negative-u charge order in N-doped WO 3 : A spin-peierls-like system Muhammad N. Huda,*, Yanfa Yan, Su-Huai Wei, and Mowafak M. Al-Jassim National Renewable Energy Laboratory, Golden,
Exchange-induced negative-u charge order in N-doped WO 3 : A spin-peierls-like system Muhammad N. Huda,*, Yanfa Yan, Su-Huai Wei, and Mowafak M. Al-Jassim National Renewable Energy Laboratory, Golden,
Ferromagnetism. Iron, nickel, and cobalt are ferromagnetic.
 Ferromagnetism Technische Universität Graz Institute of Solid State Physics Ferromagnetism elow a critical temperature (called the Curie temperature) a magnetization spontaneously appears in a ferromagnet
Ferromagnetism Technische Universität Graz Institute of Solid State Physics Ferromagnetism elow a critical temperature (called the Curie temperature) a magnetization spontaneously appears in a ferromagnet
arxiv: v2 [cond-mat.mtrl-sci] 25 Jan 2010
![arxiv: v2 [cond-mat.mtrl-sci] 25 Jan 2010](/thumbs/76/73887967.jpg "arxiv: v2 [cond-mat.mtrl-sci] 25 Jan 2010") A First-Principles Study of Zinc Oxide Honeycomb Structures arxiv:0907.3070v2 [cond-mat.mtrl-sci] 25 Jan 2010 M. Topsakal, 1 S. Cahangirov, 1 E. Bekaroglu, 1 and S. Ciraci 1, 2 1 UNAM-Institute of Materials
A First-Principles Study of Zinc Oxide Honeycomb Structures arxiv:0907.3070v2 [cond-mat.mtrl-sci] 25 Jan 2010 M. Topsakal, 1 S. Cahangirov, 1 E. Bekaroglu, 1 and S. Ciraci 1, 2 1 UNAM-Institute of Materials
Electromagnetism II. Instructor: Andrei Sirenko Spring 2013 Thursdays 1 pm 4 pm. Spring 2013, NJIT 1
 Electromagnetism II Instructor: Andrei Sirenko sirenko@njit.edu Spring 013 Thursdays 1 pm 4 pm Spring 013, NJIT 1 PROBLEMS for CH. 6 http://web.njit.edu/~sirenko/phys433/phys433eandm013.htm Can obtain
Electromagnetism II Instructor: Andrei Sirenko sirenko@njit.edu Spring 013 Thursdays 1 pm 4 pm Spring 013, NJIT 1 PROBLEMS for CH. 6 http://web.njit.edu/~sirenko/phys433/phys433eandm013.htm Can obtain
Electronic structure of solid FeO at high pressures by quantum Monte Carlo methods
 Physics Procedia 3 (2010) 1437 1441 www.elsevier.com/locate/procedia Electronic structure of solid FeO at high pressures by quantum Monte Carlo methods Jindřich Kolorenč a and Lubos Mitas a a Department
Physics Procedia 3 (2010) 1437 1441 www.elsevier.com/locate/procedia Electronic structure of solid FeO at high pressures by quantum Monte Carlo methods Jindřich Kolorenč a and Lubos Mitas a a Department
Kevin Driver 1 Shuai Zhang 1 Burkhard Militzer 1 R. E. Cohen 2.
 Quantum Monte Carlo Simulations of a Single Iron Impurity in MgO Kevin Driver 1 Shuai Zhang 1 Burkhard Militzer 1 R. E. Cohen 2 1 Department of Earth & Planetary Science University of California, Berkeley
Quantum Monte Carlo Simulations of a Single Iron Impurity in MgO Kevin Driver 1 Shuai Zhang 1 Burkhard Militzer 1 R. E. Cohen 2 1 Department of Earth & Planetary Science University of California, Berkeley