Correlation in correlated materials (mostly transition metal oxides) Lucas K. Wagner University of Illinois at Urbana-Champaign
|
|
|
- Annis Blake
- 5 years ago
- Views:
Transcription


1 Correlation in correlated materials (mostly transition metal oxides) Lucas K. Wagner University of Illinois at Urbana-Champaign
2 Understanding of correlated materials is mostly phenomenological FN- DMC (Slater- Jastrow): good energetics, but due to cancellation of errors Use reduced density matrices and accurate wave functions to calculate electron correlations
3 Transition metal oxides and agempts to describe them
4 Strongly correlated materials High Tc superconductivity Heavy fermions MoG insulators Collossal magnetoresistance Metal- insulator transitions After C. Hartinger
5 Phenomenological models of the physics: Hubbard model Valence bond theory First principles calculations: DFT (hard to include correlation) Quantum chemistry (hard to apply to solids) Quantum Monte Carlo (fixed node?) GW (perturbation theory) Phenomenological models + first principles: DFT+U DFT+DMFT
6 NiO: the band gap Experiment LDA+DMFT Jiang et al. PRB (2010) Ren et al. PRB (2006)
7 Reaction enthalpies U helps, but hard to use predictively Wang, Maxisch, and Ceder. PRB 73, (2006)
8 Problems with DFT+Hubbard: Lots of (too much) flexibility: Double- counting scheme Level of lagice model treatment Basis on which to apply lagice model How we choose the value of U Do we include intersite terms? LiGle a priori guidance! QMC methods to give guidance?
9 QMC calculations on transition metal oxide materials (a very brief summary)

10 Early work: TMO molecules E(DMC, B3LYP orbitals)-e(dmc, HF orbitals) (ev) ScO TiO VO CrO MnO σ 1 σ 1 δ 1 σ 1 δ 2 σ 1 δ 2 π 1 σ 1 δ 2 π 2 Wagner & Mitas, Chem. Phys. LeG (2003) Wagner & Mitas, J. Chem. Phys. 126, (2007)
11 FN- DMC(SJ) energetics performance is preny good General note: d- p hybridization needs to be properly described for the best results. After that, Cohesive energies in good agreement w/experiment: MnO, FeO, NiO, BaTiO 3, ZnO, V x O y Equations of state: FeO Band Gap: FeO, MnO, BaTiO 3, ZnO Needs and Towler Int. J. Mod. Phys. B (2003) Wagner, J. Phys.: Condens. MaGer (2007) Kolorenc and Mitas, Phys. Rev. LeG. 101, (2008) Kolorenc, Hu, and Mitas, Phys. Rev. B 82, (2010) Ertekin, Wagner, and Grossman, (in preparation) Bande and Luchow, Phys. Chem. Chem. Phys., 2008, 10, 3371
12 Signs of trouble: TMO molecules Bond length (angstroms) Experiment DMC LDA UCCSD(T) Binding Energy of MO (ev) Experiment DMC LDA UCCSD(T) ScO TiO VO CrO MnO ScO TiO VO CrO MnO Bond length and binding energy are very close to experiment when using B3LYP orbitals But the dipole moment still changes a lot with multiple determinants!
13 Despite encouraging energetic performance, there are indications that the wave function is not very good at the DMC(SJ) level! Going forward Guess wave functions (hope they work) General wave function expansion (but the space is exponential!) Understand the physics (too hard?)
14 Analyzing electron correlation using reduced density matrices
15 Understand the physics This has two purposes: Get beger accuracy in QMC Help understand the physics of TMO materials
16 Strong correlations? Physicists: Strong electron- electron term in effective low- energy Hamiltonian (Hubbard U) Chemists: Large static correlation (left- right correlation) Electron in place A à another electron not in place A

17 Correlation in electron gas Ĥ = 1 2 X r 2 i + X i i<j 1 r ij High density: kinetic energy dominates Low density: Coulomb energy dominates

18 Left- right correlation: H 2 molecule Electron- nucleus and kinetic energy dominate Electron- nucleus smaller, kinetic energy smaller: Electron on A à other electron on B
19 Introduction to density matrices 1(r) 2(r) Ansa^: (r 1,r 2 )=a 1 1 (r 1 ) 1 (r 2 ) a 2 2 (r 1 ) 2 (r 2 )
20 (r 1,r 2 )=a 1 1 (r 1 ) 1 (r 2 ) a 2 2 (r 1 ) 2 (r 2 ) 1RDM ij = h c i c j i 1RDM = a a 2 2 2RDM ij,ij = h c i c j c ic j i a RDM d = 0 a 2 2 c ij = 2RDM ij,ij 1RDM ii 1RDM jj c = a 2 1 a 4 1 a 2 1a 2 2 a 2 1a 2 2 a 2 2 a 4 2
21 General case (r 1,r 2 )= 1 (r 1 ) 1 (r 1 ) 1RDM = 2RDM d = a a 2 2 a a 2 2 1RDM = 2RDM d = c = a 2 1 a 4 1 a 2 1a 2 2 a 2 1a 2 2 a 2 2 a 4 2 c =
 ' 1 (r 1 ) 1 (r 2 ) c 2 (r 1 ) 2 (r 2 )")
22 2- RDM diagonal 1(r) 2(r) 2RDM d = a a 2 2 (r 1,r 2 ) ' 1 (r 1 ) 1 (r 2 ) c 2 (r 1 ) 2 (r 2 )
23 Relation to Hubbard picture If we change basis: a1 a 2 a 1 a 2 c = 1 2 a 1 a 2 a 1 a 2 c = a 2 1 a 4 1 a 2 1a 2 2 a 2 1a 2 2 a 2 2 a 4 2


24 c Positive Only on- site repulsion: More complex structure:
25 c = a 2 1 a 4 1 a 2 1a 2 2 a 2 1a 2 2 a 2 2 a 4 2 Most sets of four orbitals give rise to real- space repulsion with this pagern Including combinations of localized and diffuse orbitals. a1 a 2 a 1 a 2 c = 1 2 a 1 a 2 a 1 a 2 Localized sets of orbitals give this pagern
26 Evaluating electron correlation in transition metal oxides
27 Change in occupation number MnO 2 convergence up down ScO TiO VO CrO MnO MnO 2 FeO 2 CoO 2 Use small molecules Expand in Slater determinants, only doubles Converge the 1- RDM of the wave function with respect to determinants Cutoff threshold Orbital #


28 Electronic structure of the TM monoxides Doubly occupied Singly occupied Filling order Virtual orbitals
29 Diagonal of the 1- RDM Majority (up) Minority (down)

30 Doubly Singly Virtual orbitals Minority spin Majority spin c


31 s s dp dp dp d d d dp dp Nominal Large prob s dp
32 Localized orbitals: MnO Oxygen centered TM- centered Very clearly left- right correlation!




33 Rho^c for all 5 TM monoxides ScO TiO VO CrO MnO
 DMC(SJ) geometries: results DFT DMC(SJ) Exp MnO 2 128 140+/- 10 135+/- 5 FeO 2")
34 TM dioxides: the angle Kulik & Marzari, J. Chem. Phys. 134, (2011) DMC(SJ) geometries: results DFT DMC(SJ) Exp MnO / /- 5 FeO / /- 10 CoO /


35 TM- O2: electronic structure Singly occupied states Unoccupied states FeO2 doubly occupies CoO2 doubly occupies Doubly occupied states
")
")


36 Majority (up) Minority (down)
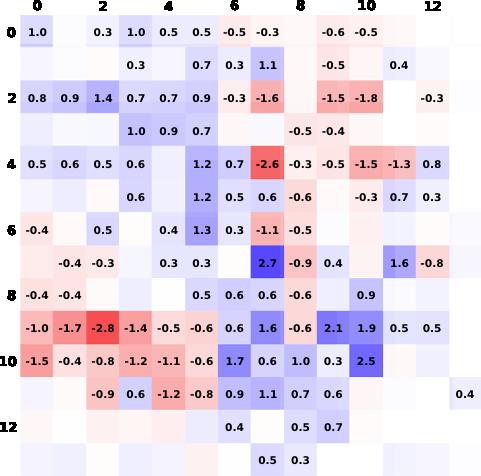
37

38 Posi tive Symmetric left- right correlation Enabled by partial d occupation
39 Special thanks: David Ceperley and his group UIUC Physics department Taub campus cluster NSF XSEDE computer resources
Quantum Monte Carlo tutorial. Lucas K. Wagner Dept. of Physics; University of Illinois at Urbana-Champaign
 Quantum Monte Carlo tutorial Lucas K. Wagner Dept. of Physics; University of Illinois at Urbana-Champaign QMC: what is it good for? Theoretical gap (ev) 5 4 3 2 1 FN-DMC DFT(PBE) VO 2 (monoclinic) La 2
Quantum Monte Carlo tutorial Lucas K. Wagner Dept. of Physics; University of Illinois at Urbana-Champaign QMC: what is it good for? Theoretical gap (ev) 5 4 3 2 1 FN-DMC DFT(PBE) VO 2 (monoclinic) La 2
Electronic structure of correlated electron systems. G.A.Sawatzky UBC Lecture
 Electronic structure of correlated electron systems G.A.Sawatzky UBC Lecture 6 011 Influence of polarizability on the crystal structure Ionic compounds are often cubic to maximize the Madelung energy i.e.
Electronic structure of correlated electron systems G.A.Sawatzky UBC Lecture 6 011 Influence of polarizability on the crystal structure Ionic compounds are often cubic to maximize the Madelung energy i.e.
New applications of Diffusion Quantum Monte Carlo
 New applications of Diffusion Quantum Monte Carlo Paul R. C. Kent (ORNL) Graphite: P. Ganesh, J. Kim, C. Park, M. Yoon, F. A. Reboredo (ORNL) Platinum: W. Parker, A. Benali, N. Romero (ANL), J. Greeley
New applications of Diffusion Quantum Monte Carlo Paul R. C. Kent (ORNL) Graphite: P. Ganesh, J. Kim, C. Park, M. Yoon, F. A. Reboredo (ORNL) Platinum: W. Parker, A. Benali, N. Romero (ANL), J. Greeley
A data-centered approach to understanding quantum behaviors in materials
 A data-centered approach to understanding quantum behaviors in materials Lucas K. Wagner Department of Physics Institute for Condensed Matter Theory National Center for Supercomputing Applications University
A data-centered approach to understanding quantum behaviors in materials Lucas K. Wagner Department of Physics Institute for Condensed Matter Theory National Center for Supercomputing Applications University
Electronic correlations in models and materials. Jan Kuneš
 Electronic correlations in models and materials Jan Kuneš Outline Dynamical-mean field theory Implementation (impurity problem) Single-band Hubbard model MnO under pressure moment collapse metal-insulator
Electronic correlations in models and materials Jan Kuneš Outline Dynamical-mean field theory Implementation (impurity problem) Single-band Hubbard model MnO under pressure moment collapse metal-insulator
Structure of CoO(001) surface from DFT+U calculations
 surface from DFT+U calculations") Structure of CoO(001) surface from DFT+U calculations B. Sitamtze Youmbi and F. Calvayrac Institut des Molécules et Matériaux du Mans (IMMM), UMR CNRS 6283 16 septembre 2013 Introduction Motivation Motivation
Structure of CoO(001) surface from DFT+U calculations B. Sitamtze Youmbi and F. Calvayrac Institut des Molécules et Matériaux du Mans (IMMM), UMR CNRS 6283 16 septembre 2013 Introduction Motivation Motivation
Band calculations: Theory and Applications
 Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
Metal-insulator transitions
 Metal-insulator transitions What can we learn from electronic structure calculations? Mike Towler mdt26@phy.cam.ac.uk www.tcm.phy.cam.ac.uk/ mdt26 Theory of Condensed Matter Group Cavendish Laboratory
Metal-insulator transitions What can we learn from electronic structure calculations? Mike Towler mdt26@phy.cam.ac.uk www.tcm.phy.cam.ac.uk/ mdt26 Theory of Condensed Matter Group Cavendish Laboratory
Principles of Quantum Mechanics
 Principles of Quantum Mechanics - indistinguishability of particles: bosons & fermions bosons: total wavefunction is symmetric upon interchange of particle coordinates (space,spin) fermions: total wavefuncftion
Principles of Quantum Mechanics - indistinguishability of particles: bosons & fermions bosons: total wavefunction is symmetric upon interchange of particle coordinates (space,spin) fermions: total wavefuncftion
An introduction to the dynamical mean-field theory. L. V. Pourovskii
 An introduction to the dynamical mean-field theory L. V. Pourovskii Nordita school on Photon-Matter interaction, Stockholm, 06.10.2016 OUTLINE The standard density-functional-theory (DFT) framework An
An introduction to the dynamical mean-field theory L. V. Pourovskii Nordita school on Photon-Matter interaction, Stockholm, 06.10.2016 OUTLINE The standard density-functional-theory (DFT) framework An
Modified Becke-Johnson (mbj) exchange potential
 exchange potential") Modified Becke-Johnson (mbj) exchange potential Hideyuki Jippo Fujitsu Laboratories LTD. 2015.12.21-22 OpenMX developer s meeting @ Kobe Overview: mbj potential The semilocal exchange potential adding
Modified Becke-Johnson (mbj) exchange potential Hideyuki Jippo Fujitsu Laboratories LTD. 2015.12.21-22 OpenMX developer s meeting @ Kobe Overview: mbj potential The semilocal exchange potential adding
arxiv: v1 [cond-mat.mtrl-sci] 21 Dec 2007
![arxiv: v1 [cond-mat.mtrl-sci] 21 Dec 2007](/thumbs/92/110189214.jpg "arxiv: v1 [cond-mat.mtrl-sci] 21 Dec 2007") Quantum Monte Carlo calculations of structural properties of FeO under pressure Jindřich Kolorenč and Lubos Mitas Department of Physics and CHiPS, North Carolina State University, Raleigh, North Carolina
Quantum Monte Carlo calculations of structural properties of FeO under pressure Jindřich Kolorenč and Lubos Mitas Department of Physics and CHiPS, North Carolina State University, Raleigh, North Carolina
Quantum Monte Carlo methods
 Quantum Monte Carlo methods Lubos Mitas North Carolina State University Urbana, August 2006 Lubos_Mitas@ncsu.edu H= 1 2 i i 2 i, I Z I r ii i j 1 r ij E ion ion H r 1, r 2,... =E r 1, r 2,... - ground
Quantum Monte Carlo methods Lubos Mitas North Carolina State University Urbana, August 2006 Lubos_Mitas@ncsu.edu H= 1 2 i i 2 i, I Z I r ii i j 1 r ij E ion ion H r 1, r 2,... =E r 1, r 2,... - ground
Magnetism in transition metal oxides by post-dft methods
 Magnetism in transition metal oxides by post-dft methods Cesare Franchini Faculty of Physics & Center for Computational Materials Science University of Vienna, Austria Workshop on Magnetism in Complex
Magnetism in transition metal oxides by post-dft methods Cesare Franchini Faculty of Physics & Center for Computational Materials Science University of Vienna, Austria Workshop on Magnetism in Complex
College of Chemistry, Peking University, Beijing, China. Fritz-Haber-Institut der MPG, Berlin, Germany
 KITP Program Excitations in Condensed Matter Localized and Itinerant States in a Unified Picture beyond Density Functional Theory Hong Jiang 1, Patrick Rinke 2 and Matthias Scheffler 2 1 College of Chemistry,
KITP Program Excitations in Condensed Matter Localized and Itinerant States in a Unified Picture beyond Density Functional Theory Hong Jiang 1, Patrick Rinke 2 and Matthias Scheffler 2 1 College of Chemistry,
Kevin Driver 1 Shuai Zhang 1 Burkhard Militzer 1 R. E. Cohen 2.
 Quantum Monte Carlo Simulations of a Single Iron Impurity in MgO Kevin Driver 1 Shuai Zhang 1 Burkhard Militzer 1 R. E. Cohen 2 1 Department of Earth & Planetary Science University of California, Berkeley
Quantum Monte Carlo Simulations of a Single Iron Impurity in MgO Kevin Driver 1 Shuai Zhang 1 Burkhard Militzer 1 R. E. Cohen 2 1 Department of Earth & Planetary Science University of California, Berkeley
Computational Exploration of Unconventional Superconductors Using Quantum Monte Carlo. Project PI: Lucas K. Wagner
 Computational Exploration of Unconventional Superconductors Using Quantum Monte Carlo Project PI: Lucas K. Wagner La2-xSrxCuO4 (LSCO) crystal structure CuO2 plane LaO spacer Peter Wahl. Nature Physics
Computational Exploration of Unconventional Superconductors Using Quantum Monte Carlo Project PI: Lucas K. Wagner La2-xSrxCuO4 (LSCO) crystal structure CuO2 plane LaO spacer Peter Wahl. Nature Physics
NiO - hole doping and bandstructure of charge transfer insulator
 NiO - hole doping and bandstructure of charge transfer insulator Jan Kuneš Institute for Physics, Uni. Augsburg Collaboration: V. I. Anisimov S. L. Skornyakov A. V. Lukoyanov D. Vollhardt Outline NiO -
NiO - hole doping and bandstructure of charge transfer insulator Jan Kuneš Institute for Physics, Uni. Augsburg Collaboration: V. I. Anisimov S. L. Skornyakov A. V. Lukoyanov D. Vollhardt Outline NiO -
Electronic structure of correlated electron systems. Lecture 2
 Electronic structure of correlated electron systems Lecture 2 Band Structure approach vs atomic Band structure Delocalized Bloch states Fill up states with electrons starting from the lowest energy No
Electronic structure of correlated electron systems Lecture 2 Band Structure approach vs atomic Band structure Delocalized Bloch states Fill up states with electrons starting from the lowest energy No
v(r i r j ) = h(r i )+ 1 N
 = h(r i )+ 1 N") Chapter 1 Hartree-Fock Theory 1.1 Formalism For N electrons in an external potential V ext (r), the many-electron Hamiltonian can be written as follows: N H = [ p i i=1 m +V ext(r i )]+ 1 N N v(r i r j
Chapter 1 Hartree-Fock Theory 1.1 Formalism For N electrons in an external potential V ext (r), the many-electron Hamiltonian can be written as follows: N H = [ p i i=1 m +V ext(r i )]+ 1 N N v(r i r j
Ab-initio molecular dynamics for High pressure Hydrogen
 Ab-initio molecular dynamics for High pressure Hydrogen Claudio Attaccalite Institut d'electronique, Microélectronique et Nanotechnologie (IEMN), Lille Outline A brief introduction to Quantum Monte Carlo
Ab-initio molecular dynamics for High pressure Hydrogen Claudio Attaccalite Institut d'electronique, Microélectronique et Nanotechnologie (IEMN), Lille Outline A brief introduction to Quantum Monte Carlo
Quantum Mechanical Simulations
 Quantum Mechanical Simulations Prof. Yan Wang Woodruff School of Mechanical Engineering Georgia Institute of Technology Atlanta, GA 30332, U.S.A. yan.wang@me.gatech.edu Topics Quantum Monte Carlo Hartree-Fock
Quantum Mechanical Simulations Prof. Yan Wang Woodruff School of Mechanical Engineering Georgia Institute of Technology Atlanta, GA 30332, U.S.A. yan.wang@me.gatech.edu Topics Quantum Monte Carlo Hartree-Fock
Variational Monte Carlo Optimization and Excited States
 Variational Monte Carlo Optimization and Excited States Eric Neuscamman August 9, 2018 motivation charge transfer core spectroscopy double excitations the menu aperitif: number counting Jastrows main course:
Variational Monte Carlo Optimization and Excited States Eric Neuscamman August 9, 2018 motivation charge transfer core spectroscopy double excitations the menu aperitif: number counting Jastrows main course:
w2dynamics : operation and applications
 w2dynamics : operation and applications Giorgio Sangiovanni ERC Kick-off Meeting, 2.9.2013 Hackers Nico Parragh (Uni Wü) Markus Wallerberger (TU) Patrik Gunacker (TU) Andreas Hausoel (Uni Wü) A solver
w2dynamics : operation and applications Giorgio Sangiovanni ERC Kick-off Meeting, 2.9.2013 Hackers Nico Parragh (Uni Wü) Markus Wallerberger (TU) Patrik Gunacker (TU) Andreas Hausoel (Uni Wü) A solver
Introduction to Density Functional Theory
 1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
Auxiliary-field quantum Monte Carlo calculations of excited states and strongly correlated systems
 Auxiliary-field quantum Monte Carlo calculations of excited states and strongly correlated systems Formally simple -- a framework for going beyond DFT? Random walks in non-orthogonal Slater determinant
Auxiliary-field quantum Monte Carlo calculations of excited states and strongly correlated systems Formally simple -- a framework for going beyond DFT? Random walks in non-orthogonal Slater determinant
Electronic correlation and Hubbard approaches
 Electronic correlation and Hubbard approaches Matteo Cococcioni Department of Chemical Engineering and Materials Science University of Minnesota Notable failures of LDA/GGA: transition-metal oxides Introduction
Electronic correlation and Hubbard approaches Matteo Cococcioni Department of Chemical Engineering and Materials Science University of Minnesota Notable failures of LDA/GGA: transition-metal oxides Introduction
First- principles studies of spin-crossover molecules
 Vallico di Sotto, 30 th July 2012 First- principles studies of spin-crossover molecules Andrea Droghetti and Stefano Sanvito School of Physics and CRANN, Trinity College Dublin Dario Alfe' London Centre
Vallico di Sotto, 30 th July 2012 First- principles studies of spin-crossover molecules Andrea Droghetti and Stefano Sanvito School of Physics and CRANN, Trinity College Dublin Dario Alfe' London Centre
Theoretical study of electronic and atomic structures of (MnO)n
n") Theoretical study of electronic and atomic structures of (MnO)n Hiori Kino, a Lucas K. Wagner b and Lubos Mitas c a National Institute for Materials Science, 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan.
Theoretical study of electronic and atomic structures of (MnO)n Hiori Kino, a Lucas K. Wagner b and Lubos Mitas c a National Institute for Materials Science, 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan.
OVERVIEW OF QUANTUM CHEMISTRY METHODS
 OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
Quantum Monte Carlo for Transition Metal Systems: Method Developments and Applications. Lucas K. Wagner
 ABSTRACT WAGNER, LUCAS K. Quantum Monte Carlo for Transition Metal Systems: Method Developments and Applications. (Under the direction of Professor Lubos Mitas). Quantum Monte Carlo (QMC) is a powerful
ABSTRACT WAGNER, LUCAS K. Quantum Monte Carlo for Transition Metal Systems: Method Developments and Applications. (Under the direction of Professor Lubos Mitas). Quantum Monte Carlo (QMC) is a powerful
MO Calculation for a Diatomic Molecule. /4 0 ) i=1 j>i (1/r ij )
 i=1 j>i (1/r ij )") MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
Introduction to Computational Chemistry
 Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Mott insulators. Mott-Hubbard type vs charge-transfer type
 Mott insulators Mott-Hubbard type vs charge-transfer type Cluster-model description Chemical trend Band theory Self-energy correction Electron-phonon interaction Mott insulators Mott-Hubbard type vs charge-transfer
Mott insulators Mott-Hubbard type vs charge-transfer type Cluster-model description Chemical trend Band theory Self-energy correction Electron-phonon interaction Mott insulators Mott-Hubbard type vs charge-transfer
Dynamical mean-field theory and strong correlations in solids and molecules
 Dynamical mean-field theory and strong correlations in solids and molecules Collaborators: Cedric Weber Peter B. Littlewood Mike C. Payne Gabriel Kotliar David D. O Regan Nicholas D. M. Hine 1 Outlines
Dynamical mean-field theory and strong correlations in solids and molecules Collaborators: Cedric Weber Peter B. Littlewood Mike C. Payne Gabriel Kotliar David D. O Regan Nicholas D. M. Hine 1 Outlines
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that. Keith Refson STFC Rutherford Appleton Laboratory
 Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
Electronic band structure, sx-lda, Hybrid DFT, LDA+U and all that Keith Refson STFC Rutherford Appleton Laboratory LDA/GGA DFT is good but... Naive LDA/GGA calculation severely underestimates band-gaps.
Exchange-Correlation Functional
 Exchange-Correlation Functional Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Exchange-Correlation Functional Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Quantum Monte Carlo Simulations of Exciton Condensates
 Quantum Monte Carlo Simulations of Exciton Condensates J. Shumway a and D. M. Ceperley b a Dept. of Physics and Astronomy, Arizona State University, Tempe, AZ 8583 b Dept. of Physics, University of Illinois,
Quantum Monte Carlo Simulations of Exciton Condensates J. Shumway a and D. M. Ceperley b a Dept. of Physics and Astronomy, Arizona State University, Tempe, AZ 8583 b Dept. of Physics, University of Illinois,
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić
 Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Crystalline and Magnetic Anisotropy of the 3d Transition-Metal Monoxides
 Crystalline and of the 3d Transition-Metal Monoxides Institut für Festkörpertheorie und -optik Friedrich-Schiller-Universität Max-Wien-Platz 1 07743 Jena 2012-03-23 Introduction Crystalline Anisotropy
Crystalline and of the 3d Transition-Metal Monoxides Institut für Festkörpertheorie und -optik Friedrich-Schiller-Universität Max-Wien-Platz 1 07743 Jena 2012-03-23 Introduction Crystalline Anisotropy
Magnetism and Superconductivity in Decorated Lattices
 Magnetism and Superconductivity in Decorated Lattices Mott Insulators and Antiferromagnetism- The Hubbard Hamiltonian Illustration: The Square Lattice Bipartite doesn t mean N A = N B : The Lieb Lattice
Magnetism and Superconductivity in Decorated Lattices Mott Insulators and Antiferromagnetism- The Hubbard Hamiltonian Illustration: The Square Lattice Bipartite doesn t mean N A = N B : The Lieb Lattice
Beyond the Hartree-Fock Approximation: Configuration Interaction
 Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
Introduction and Overview of the Reduced Density Matrix Functional Theory
 Introduction and Overview of the Reduced Density Matrix Functional Theory N. N. Lathiotakis Theoretical and Physical Chemistry Institute, National Hellenic Research Foundation, Athens April 13, 2016 Outline
Introduction and Overview of the Reduced Density Matrix Functional Theory N. N. Lathiotakis Theoretical and Physical Chemistry Institute, National Hellenic Research Foundation, Athens April 13, 2016 Outline
QMC dissociation energy of the water dimer: Time step errors and backflow calculations
 QMC dissociation energy of the water dimer: Time step errors and backflow calculations Idoia G. de Gurtubay and Richard J. Needs TCM group. Cavendish Laboratory University of Cambridge Idoia G. de Gurtubay.
QMC dissociation energy of the water dimer: Time step errors and backflow calculations Idoia G. de Gurtubay and Richard J. Needs TCM group. Cavendish Laboratory University of Cambridge Idoia G. de Gurtubay.
All-electron quantum Monte Carlo calculations for the noble gas atoms He to Xe
 All-electron quantum Monte Carlo calculations for the noble gas atoms He to Xe A. Ma, N. D. Drummond, M. D. Towler, and R. J. Needs Theory of Condensed Matter Group, Cavendish Laboratory, University of
All-electron quantum Monte Carlo calculations for the noble gas atoms He to Xe A. Ma, N. D. Drummond, M. D. Towler, and R. J. Needs Theory of Condensed Matter Group, Cavendish Laboratory, University of
Quantum Monte Carlo calculations of medium mass nuclei
 Quantum Monte Carlo calculations of medium mass nuclei Diego Lonardoni FRIB Theory Fellow In collaboration with: J. Carlson, LANL S. Gandolfi, LANL X. Wang, Huzhou University, China A. Lovato, ANL & UniTN
Quantum Monte Carlo calculations of medium mass nuclei Diego Lonardoni FRIB Theory Fellow In collaboration with: J. Carlson, LANL S. Gandolfi, LANL X. Wang, Huzhou University, China A. Lovato, ANL & UniTN
Journal of Theoretical Physics
 1 Journal of Theoretical Physics Founded and Edited by M. Apostol 53 (2000) ISSN 1453-4428 Ionization potential for metallic clusters L. C. Cune and M. Apostol Department of Theoretical Physics, Institute
1 Journal of Theoretical Physics Founded and Edited by M. Apostol 53 (2000) ISSN 1453-4428 Ionization potential for metallic clusters L. C. Cune and M. Apostol Department of Theoretical Physics, Institute
Luigi Paolasini
 Luigi Paolasini paolasini@esrf.fr LECTURE 4: MAGNETIC INTERACTIONS - Dipole vs exchange magnetic interactions. - Direct and indirect exchange interactions. - Anisotropic exchange interactions. - Interplay
Luigi Paolasini paolasini@esrf.fr LECTURE 4: MAGNETIC INTERACTIONS - Dipole vs exchange magnetic interactions. - Direct and indirect exchange interactions. - Anisotropic exchange interactions. - Interplay
Fixed-Node quantum Monte Carlo for Chemistry
 Fixed-Node quantum Monte Carlo for Chemistry Michel Caffarel Lab. Physique et Chimie Quantiques, CNRS-IRSAMC, Université de Toulouse e-mail : caffarel@irsamc.ups-tlse.fr. p.1/29 The N-body problem of Chemistry
Fixed-Node quantum Monte Carlo for Chemistry Michel Caffarel Lab. Physique et Chimie Quantiques, CNRS-IRSAMC, Université de Toulouse e-mail : caffarel@irsamc.ups-tlse.fr. p.1/29 The N-body problem of Chemistry
Magnetic Moment Collapse drives Mott transition in MnO
 Magnetic Moment Collapse drives Mott transition in MnO J. Kuneš Institute of Physics, Uni. Augsburg in collaboration with: V. I. Anisimov, A. V. Lukoyanov, W. E. Pickett, R. T. Scalettar, D. Vollhardt,
Magnetic Moment Collapse drives Mott transition in MnO J. Kuneš Institute of Physics, Uni. Augsburg in collaboration with: V. I. Anisimov, A. V. Lukoyanov, W. E. Pickett, R. T. Scalettar, D. Vollhardt,
X-ray absorption spectroscopy.
 X-ray absorption spectroscopy www.anorg.chem.uu.nl/people/staff/frankdegroot/ X-ray absorption spectroscopy www.anorg.chem.uu.nl/people/staff/frankdegroot/ Frank de Groot PhD: solid state chemistry U Nijmegen
X-ray absorption spectroscopy www.anorg.chem.uu.nl/people/staff/frankdegroot/ X-ray absorption spectroscopy www.anorg.chem.uu.nl/people/staff/frankdegroot/ Frank de Groot PhD: solid state chemistry U Nijmegen
Lec20 Fri 3mar17
 564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
Electronic structure quantum Monte Carlo: pfaffians and many-body nodes of ground and excited states
 Electronic structure quantum Monte Carlo: pfaffians and many-body nodes of ground and excited states Jindrich Kolorenc (von Humboldt Fellow), U. Hamburg Michal Bajdich, ORNL Lubos Mitas, North Carolina
Electronic structure quantum Monte Carlo: pfaffians and many-body nodes of ground and excited states Jindrich Kolorenc (von Humboldt Fellow), U. Hamburg Michal Bajdich, ORNL Lubos Mitas, North Carolina
Electronic structure of solid FeO at high pressures by quantum Monte Carlo methods
 Physics Procedia 3 (2010) 1437 1441 www.elsevier.com/locate/procedia Electronic structure of solid FeO at high pressures by quantum Monte Carlo methods Jindřich Kolorenč a and Lubos Mitas a a Department
Physics Procedia 3 (2010) 1437 1441 www.elsevier.com/locate/procedia Electronic structure of solid FeO at high pressures by quantum Monte Carlo methods Jindřich Kolorenč a and Lubos Mitas a a Department
Rh 3d. Co 2p. Binding Energy (ev) Binding Energy (ev) (b) (a)
 Binding Energy (ev) (b) (a)") Co 2p Co(0) 778.3 Rh 3d Rh (0) 307.2 810 800 790 780 770 Binding Energy (ev) (a) 320 315 310 305 Binding Energy (ev) (b) Supplementary Figure 1 Photoemission features of a catalyst precursor which was
Co 2p Co(0) 778.3 Rh 3d Rh (0) 307.2 810 800 790 780 770 Binding Energy (ev) (a) 320 315 310 305 Binding Energy (ev) (b) Supplementary Figure 1 Photoemission features of a catalyst precursor which was
Lecture 19: Building Atoms and Molecules
 Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r y even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in
Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r y even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in
Diagrammatic Monte Carlo methods for Fermions
 Diagrammatic Monte Carlo methods for Fermions Philipp Werner Department of Physics, Columbia University PRL 97, 7645 (26) PRB 74, 15517 (26) PRB 75, 8518 (27) PRB 76, 235123 (27) PRL 99, 12645 (27) PRL
Diagrammatic Monte Carlo methods for Fermions Philipp Werner Department of Physics, Columbia University PRL 97, 7645 (26) PRB 74, 15517 (26) PRB 75, 8518 (27) PRB 76, 235123 (27) PRL 99, 12645 (27) PRL
Quantum Monte Carlo methods: recent developments and applications
 Quantum Monte Carlo methods: recent developments and applications Lucas Wagner, Michal Bajdich, Gabriel Drobny Zack Helms, Lubos Mitas North Carolina State University in collab. with Jeffrey Grossman,
Quantum Monte Carlo methods: recent developments and applications Lucas Wagner, Michal Bajdich, Gabriel Drobny Zack Helms, Lubos Mitas North Carolina State University in collab. with Jeffrey Grossman,
QUANTUM CHEMISTRY FOR TRANSITION METALS
 QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
Bayero Journal of Pure and Applied Sciences, 3(1): Received: September, 2009 Accepted: May, 2010
: Received: September, 2009 Accepted: May, 2010") Bajopas Volume 3 Number June Bayero Journal of Pure and Applied Sciences, 3(: - 7 Received: September, 9 Accepted: May, VARIAIONAL QUANUM MON CARLO CALCULAION OF H GROUND SA NRG OF HDROGN MOLCUL Suleiman,
Bajopas Volume 3 Number June Bayero Journal of Pure and Applied Sciences, 3(: - 7 Received: September, 9 Accepted: May, VARIAIONAL QUANUM MON CARLO CALCULAION OF H GROUND SA NRG OF HDROGN MOLCUL Suleiman,
arxiv: v1 [cond-mat.str-el] 18 Jul 2007
![arxiv: v1 [cond-mat.str-el] 18 Jul 2007](/thumbs/89/99838639.jpg "arxiv: v1 [cond-mat.str-el] 18 Jul 2007") arxiv:0707.2704v1 [cond-mat.str-el] 18 Jul 2007 Determination of the Mott insulating transition by the multi-reference density functional theory 1. Introduction K. Kusakabe Graduate School of Engineering
arxiv:0707.2704v1 [cond-mat.str-el] 18 Jul 2007 Determination of the Mott insulating transition by the multi-reference density functional theory 1. Introduction K. Kusakabe Graduate School of Engineering
The Gutzwiller Density Functional Theory
 The Gutzwiller Density Functional Theory Jörg Bünemann, BTU Cottbus I) Introduction 1. Model for an H 2 -molecule 2. Transition metals and their compounds II) Gutzwiller variational theory 1. Gutzwiller
The Gutzwiller Density Functional Theory Jörg Bünemann, BTU Cottbus I) Introduction 1. Model for an H 2 -molecule 2. Transition metals and their compounds II) Gutzwiller variational theory 1. Gutzwiller
LUMO + 1 LUMO. Tómas Arnar Guðmundsson Report 2 Reikniefnafræði G
 Q1: Display all the MOs for N2 in your report and classify each one of them as bonding, antibonding or non-bonding, and say whether the symmetry of the orbital is σ or π. Sketch a molecular orbital diagram
Q1: Display all the MOs for N2 in your report and classify each one of them as bonding, antibonding or non-bonding, and say whether the symmetry of the orbital is σ or π. Sketch a molecular orbital diagram
The Schrödinger equation for many-electron systems
 The Schrödinger equation for many-electron systems Ĥ!( x,, x ) = E!( x,, x ) 1 N 1 1 Z 1 Ĥ = " $ # " $ + $ 2 r 2 A j j A, j RAj i, j < i a linear differential equation in 4N variables (atomic units) (3
The Schrödinger equation for many-electron systems Ĥ!( x,, x ) = E!( x,, x ) 1 N 1 1 Z 1 Ĥ = " $ # " $ + $ 2 r 2 A j j A, j RAj i, j < i a linear differential equation in 4N variables (atomic units) (3
The Overhauser Instability
 The Overhauser Instability Zoltán Radnai and Richard Needs TCM Group ESDG Talk 14th February 2007 Typeset by FoilTEX Introduction Hartree-Fock theory and Homogeneous Electron Gas Noncollinear spins and
The Overhauser Instability Zoltán Radnai and Richard Needs TCM Group ESDG Talk 14th February 2007 Typeset by FoilTEX Introduction Hartree-Fock theory and Homogeneous Electron Gas Noncollinear spins and
Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory
 Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley National
Electronic structure theory: Fundamentals to frontiers. 1. Hartree-Fock theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley National
Computational strongly correlated materials R. Torsten Clay Physics & Astronomy
 Computational strongly correlated materials R. Torsten Clay Physics & Astronomy Current/recent students Saurabh Dayal (current PhD student) Wasanthi De Silva (new grad student 212) Jeong-Pil Song (finished
Computational strongly correlated materials R. Torsten Clay Physics & Astronomy Current/recent students Saurabh Dayal (current PhD student) Wasanthi De Silva (new grad student 212) Jeong-Pil Song (finished
CHEM3023: Spins, Atoms and Molecules
 CHEM3023: Spins, Atoms and Molecules Lecture 4 Molecular orbitals C.-K. Skylaris Learning outcomes Be able to manipulate expressions involving spin orbitals and molecular orbitals Be able to write down
CHEM3023: Spins, Atoms and Molecules Lecture 4 Molecular orbitals C.-K. Skylaris Learning outcomes Be able to manipulate expressions involving spin orbitals and molecular orbitals Be able to write down
Multiconfigurational Quantum Chemistry. Björn O. Roos as told by RL Department of Theoretical Chemistry Chemical Center Lund University Sweden
 Multiconfigurational Quantum Chemistry Björn O. Roos as told by RL Department of Theoretical Chemistry Chemical Center Lund University Sweden April 20, 2009 1 The Slater determinant Using the spin-orbitals,
Multiconfigurational Quantum Chemistry Björn O. Roos as told by RL Department of Theoretical Chemistry Chemical Center Lund University Sweden April 20, 2009 1 The Slater determinant Using the spin-orbitals,
Agilent 8800 Triple Quadrupole ICP-MS: Understanding oxygen reaction mode in ICP-MS/MS
 Agilent 8800 Triple Quadrupole ICP-MS: Understanding oxygen reaction mode in ICP-MS/MS Agilent 8800 ICP-QQQ Technical Overview Introduction The use of oxygen as a reaction gas for the removal of interferences
Agilent 8800 Triple Quadrupole ICP-MS: Understanding oxygen reaction mode in ICP-MS/MS Agilent 8800 ICP-QQQ Technical Overview Introduction The use of oxygen as a reaction gas for the removal of interferences
Module 6 1. Density functional theory
 Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
Chem 4502 Introduction to Quantum Mechanics and Spectroscopy 3 Credits Fall Semester 2014 Laura Gagliardi. Lecture 28, December 08, 2014
 Chem 4502 Introduction to Quantum Mechanics and Spectroscopy 3 Credits Fall Semester 2014 Laura Gagliardi Lecture 28, December 08, 2014 Solved Homework Water, H 2 O, involves 2 hydrogen atoms and an oxygen
Chem 4502 Introduction to Quantum Mechanics and Spectroscopy 3 Credits Fall Semester 2014 Laura Gagliardi Lecture 28, December 08, 2014 Solved Homework Water, H 2 O, involves 2 hydrogen atoms and an oxygen
Answers Quantum Chemistry NWI-MOL406 G. C. Groenenboom and G. A. de Wijs, HG00.307, 8:30-11:30, 21 jan 2014
 Answers Quantum Chemistry NWI-MOL406 G. C. Groenenboom and G. A. de Wijs, HG00.307, 8:30-11:30, 21 jan 2014 Question 1: Basis sets Consider the split valence SV3-21G one electron basis set for formaldehyde
Answers Quantum Chemistry NWI-MOL406 G. C. Groenenboom and G. A. de Wijs, HG00.307, 8:30-11:30, 21 jan 2014 Question 1: Basis sets Consider the split valence SV3-21G one electron basis set for formaldehyde
2 B B D (E) Paramagnetic Susceptibility. m s probability. A) Bound Electrons in Atoms
 Paramagnetic Susceptibility. m s probability. A) Bound Electrons in Atoms") Paramagnetic Susceptibility A) Bound Electrons in Atoms m s probability B +½ p ½e x Curie Law: 1/T s=½ + B ½ p + ½e +x With increasing temperature T the alignment of the magnetic moments in a B field is
Paramagnetic Susceptibility A) Bound Electrons in Atoms m s probability B +½ p ½e x Curie Law: 1/T s=½ + B ½ p + ½e +x With increasing temperature T the alignment of the magnetic moments in a B field is
NCTS One-Day Workshop. Yasutami Takada. cf. YT, Ryo Maezono, & Kanako Yoshizawa, Phys. Rev. B 92, (2015)
") NCTS One-Day Workshop Emergence of a spin-singlet resonance state with Kondo temperature well beyond 1000K in the proton-embedded electron gas: New route to room-temperature superconductivity Yasutami
NCTS One-Day Workshop Emergence of a spin-singlet resonance state with Kondo temperature well beyond 1000K in the proton-embedded electron gas: New route to room-temperature superconductivity Yasutami
CHEM6085: Density Functional Theory Lecture 10
 CHEM6085: Density Functional Theory Lecture 10 1) Spin-polarised calculations 2) Geometry optimisation C.-K. Skylaris 1 Unpaired electrons So far we have developed Kohn-Sham DFT for the case of paired
CHEM6085: Density Functional Theory Lecture 10 1) Spin-polarised calculations 2) Geometry optimisation C.-K. Skylaris 1 Unpaired electrons So far we have developed Kohn-Sham DFT for the case of paired
Quantum Monte Carlo wave functions and their optimization for quantum chemistry
 Quantum Monte Carlo wave functions and their optimization for quantum chemistry Julien Toulouse Université Pierre & Marie Curie and CNRS, Paris, France CEA Saclay, SPhN Orme des Merisiers April 2015 Outline
Quantum Monte Carlo wave functions and their optimization for quantum chemistry Julien Toulouse Université Pierre & Marie Curie and CNRS, Paris, France CEA Saclay, SPhN Orme des Merisiers April 2015 Outline
CHEM3023: Spins, Atoms and Molecules
 CHEM3023: Spins, Atoms and Molecules Lecture 5 The Hartree-Fock method C.-K. Skylaris Learning outcomes Be able to use the variational principle in quantum calculations Be able to construct Fock operators
CHEM3023: Spins, Atoms and Molecules Lecture 5 The Hartree-Fock method C.-K. Skylaris Learning outcomes Be able to use the variational principle in quantum calculations Be able to construct Fock operators
Dept of Mechanical Engineering MIT Nanoengineering group
 1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
Valence bond theory accounts, at least qualitatively, for the stability of the covalent bond in terms of overlapping atomic orbitals.
 Molecular Orbital Theory Valence bond theory accounts, at least qualitatively, for the stability of the covalent bond in terms of overlapping atomic orbitals. Using the concept of hybridization, valence
Molecular Orbital Theory Valence bond theory accounts, at least qualitatively, for the stability of the covalent bond in terms of overlapping atomic orbitals. Using the concept of hybridization, valence
Electronic structure quantum Monte Carlo methods and variable spins: beyond fixedphase/node
 Electronic structure quantum Monte Carlo methods and variable spins: beyond fixedphase/node approximations Cody Melton, M. Chandler Bennett, L. Mitas, with A. Ambrosetti, F. Pederiva North Carolina State
Electronic structure quantum Monte Carlo methods and variable spins: beyond fixedphase/node approximations Cody Melton, M. Chandler Bennett, L. Mitas, with A. Ambrosetti, F. Pederiva North Carolina State
Preface Introduction to the electron liquid
 Table of Preface page xvii 1 Introduction to the electron liquid 1 1.1 A tale of many electrons 1 1.2 Where the electrons roam: physical realizations of the electron liquid 5 1.2.1 Three dimensions 5 1.2.2
Table of Preface page xvii 1 Introduction to the electron liquid 1 1.1 A tale of many electrons 1 1.2 Where the electrons roam: physical realizations of the electron liquid 5 1.2.1 Three dimensions 5 1.2.2
Lecture 9. Hartree Fock Method and Koopman s Theorem
 Lecture 9 Hartree Fock Method and Koopman s Theorem Ψ(N) is approximated as a single slater determinant Φ of N orthogonal One electron spin-orbitals. One electron orbital φ i = φ i (r) χ i (σ) χ i (σ)
Lecture 9 Hartree Fock Method and Koopman s Theorem Ψ(N) is approximated as a single slater determinant Φ of N orthogonal One electron spin-orbitals. One electron orbital φ i = φ i (r) χ i (σ) χ i (σ)
The Nuclear Many-Body Problem. Lecture 2
 The Nuclear Many-Body Problem Lecture 2 How do we describe nuclei? Shell structure in nuclei and the phenomenological shell model approach to nuclear structure. Ab-initio approach to nuclear structure.
The Nuclear Many-Body Problem Lecture 2 How do we describe nuclei? Shell structure in nuclei and the phenomenological shell model approach to nuclear structure. Ab-initio approach to nuclear structure.
Spin-crossover molecules: puzzling systems for electronic structure methods. Andrea Droghetti School of Physics and CRANN, Trinity College Dublin
 Spin-crossover molecules: puzzling systems for electronic structure methods Andrea Droghetti School of Physics and CRANN, Trinity College Dublin Introduction Introduction Can we read a molecule spin? Can
Spin-crossover molecules: puzzling systems for electronic structure methods Andrea Droghetti School of Physics and CRANN, Trinity College Dublin Introduction Introduction Can we read a molecule spin? Can
Faddeev Random Phase Approximation (FRPA) Application to Molecules
 Application to Molecules") Faddeev Random Phase Approximation (FRPA) Application to Molecules Matthias Degroote Center for Molecular Modeling (CMM) Ghent University INT 2011 Spring Program Fermions from Cold Atoms to Neutron Stars:
Faddeev Random Phase Approximation (FRPA) Application to Molecules Matthias Degroote Center for Molecular Modeling (CMM) Ghent University INT 2011 Spring Program Fermions from Cold Atoms to Neutron Stars:
Electronic structure calculations results from LDA+U method
 Electronic structure calculations results from LDA+U method Vladimir I. Anisimov Institute of Metal Physics Ekaterinburg, Russia LDA+U method applications Mott insulators Polarons and stripes in cuprates
Electronic structure calculations results from LDA+U method Vladimir I. Anisimov Institute of Metal Physics Ekaterinburg, Russia LDA+U method applications Mott insulators Polarons and stripes in cuprates
ATOMIC STRUCTURE. Atomic Structure. Atomic orbitals and their energies (a) Hydrogenic radial wavefunctions
 Hydrogenic radial wavefunctions") ATOMIC STRUCTURE Atomic orbitals and their energies (a) Hydrogenic radial wavefunctions Bundet Boekfa Chem Div, Fac Lib Arts & Sci Kasetsart University Kamphaeng Saen Campus 1 2 Atomic orbitals and their
ATOMIC STRUCTURE Atomic orbitals and their energies (a) Hydrogenic radial wavefunctions Bundet Boekfa Chem Div, Fac Lib Arts & Sci Kasetsart University Kamphaeng Saen Campus 1 2 Atomic orbitals and their
Strong Correlation Effects in Fullerene Molecules and Solids
 Strong Correlation Effects in Fullerene Molecules and Solids Fei Lin Physics Department, Virginia Tech, Blacksburg, VA 2461 Fei Lin (Virginia Tech) Correlations in Fullerene SESAPS 211, Roanoke, VA 1 /
Strong Correlation Effects in Fullerene Molecules and Solids Fei Lin Physics Department, Virginia Tech, Blacksburg, VA 2461 Fei Lin (Virginia Tech) Correlations in Fullerene SESAPS 211, Roanoke, VA 1 /
Quasiparticle dynamics and interactions in non uniformly polarizable solids
 Quasiparticle dynamics and interactions in non uniformly polarizable solids Mona Berciu University of British Columbia à beautiful physics that George Sawatzky has been pursuing for a long time à today,
Quasiparticle dynamics and interactions in non uniformly polarizable solids Mona Berciu University of British Columbia à beautiful physics that George Sawatzky has been pursuing for a long time à today,
CHEM J-5 June 2014
 CHEM1101 2014-J-5 June 2014 The molecular orbital energy level diagrams for H 2, H 2 +, H 2 and O 2 are shown below. Fill in the valence electrons for each species in its ground state and label the types
CHEM1101 2014-J-5 June 2014 The molecular orbital energy level diagrams for H 2, H 2 +, H 2 and O 2 are shown below. Fill in the valence electrons for each species in its ground state and label the types
Introduction to Heisenberg model. Javier Junquera
 Introduction to Heisenberg model Javier Junquera Most important reference followed in this lecture Magnetism in Condensed Matter Physics Stephen Blundell Oxford Master Series in Condensed Matter Physics
Introduction to Heisenberg model Javier Junquera Most important reference followed in this lecture Magnetism in Condensed Matter Physics Stephen Blundell Oxford Master Series in Condensed Matter Physics
Electric properties of molecules
 Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
The Peierls distortion seen in 1D chains: The simplest model for a gap.
 The Peierls distortion seen in 1D chains: The simplest model for a gap. fold back halve distort E k Note that we go from being valence-imprecise to being valence precise: Now two electrons per unit cell.
The Peierls distortion seen in 1D chains: The simplest model for a gap. fold back halve distort E k Note that we go from being valence-imprecise to being valence precise: Now two electrons per unit cell.
ELECTRONIC STRUCTURE OF MAGNESIUM OXIDE
 Int. J. Chem. Sci.: 8(3), 2010, 1749-1756 ELECTRONIC STRUCTURE OF MAGNESIUM OXIDE P. N. PIYUSH and KANCHAN LATA * Department of Chemistry, B. N. M. V. College, Sahugarh, MADHIPUR (Bihar) INDIA ABSTRACT
Int. J. Chem. Sci.: 8(3), 2010, 1749-1756 ELECTRONIC STRUCTURE OF MAGNESIUM OXIDE P. N. PIYUSH and KANCHAN LATA * Department of Chemistry, B. N. M. V. College, Sahugarh, MADHIPUR (Bihar) INDIA ABSTRACT
Lecture 6. Fermion Pairing. WS2010/11: Introduction to Nuclear and Particle Physics
 Lecture 6 Fermion Pairing WS2010/11: Introduction to Nuclear and Particle Physics Experimental indications for Cooper-Pairing Solid state physics: Pairing of electrons near the Fermi surface with antiparallel
Lecture 6 Fermion Pairing WS2010/11: Introduction to Nuclear and Particle Physics Experimental indications for Cooper-Pairing Solid state physics: Pairing of electrons near the Fermi surface with antiparallel
the abdus salam international centre for theoretical physics
 united nations educational, scientific and cultural organization the abdus salam international centre for theoretical physics international atomic energy agency SMR 1595-16 Joint DEMOCRITOS - ICTP School
united nations educational, scientific and cultural organization the abdus salam international centre for theoretical physics international atomic energy agency SMR 1595-16 Joint DEMOCRITOS - ICTP School
CHEMISTRY 4021/8021 MIDTERM EXAM 1 SPRING 2014
 CHEMISTRY 4021/8021 Q1) Propose a simple, united-atom molecular mechanics force-field needed to generate a potential energy surface for an isolated molecule of acetone (Me 2 CO). I.e., provide an energy
CHEMISTRY 4021/8021 Q1) Propose a simple, united-atom molecular mechanics force-field needed to generate a potential energy surface for an isolated molecule of acetone (Me 2 CO). I.e., provide an energy
Section 3 Electronic Configurations, Term Symbols, and States
 Section 3 Electronic Configurations, Term Symbols, and States Introductory Remarks- The Orbital, Configuration, and State Pictures of Electronic Structure One of the goals of quantum chemistry is to allow
Section 3 Electronic Configurations, Term Symbols, and States Introductory Remarks- The Orbital, Configuration, and State Pictures of Electronic Structure One of the goals of quantum chemistry is to allow
Lecture 19: Building Atoms and Molecules
 Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r ψ even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in
Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r ψ even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in