arxiv:mtrl-th/ v1 2 Sep 1996
|
|
|
- Asher Bell
- 5 years ago
- Views:
Transcription
1 arxiv:mtrl-th/ v1 2 Sep 1996 Correlation effects in MgO and CaO: Cohesive energies and lattice constants Klaus Doll, Michael Dolg Max-Planck-Institut für Physik komplexer Systeme D Dresden, Germany Hermann Stoll Institut für Theoretische Chemie Universität Stuttgart D Stuttgart, Germany October 31, 2018 Abstract A recently proposed computational scheme based on local increments has been applied to the calculation of correlation contributions to the cohesive energy of the CaO crystal. Using ab-initio quantum chemical methods for evaluating individual increments, we obtain 80% of the difference between the experimental and Hartree-Fock cohesive energies. Lattice constants corrected for correlation effects deviate by less than 1% from experimental values, in the case of MgO and CaO. accepted by Phys. Rev. B 0
2 1 Introduction Ab-initio Hartree-Fock (HF) and configuration interaction (CI) methods are standard tools in computational chemistry nowadays and various program packages are available for accurate calculations of properties of atoms and molecules. For solids, HF calculations have become possible, on a broad scale, with the advent of the program package CRYSTAL [1]. However, the problem of an accurate treatment of electron correlation is not fully settled (for a survey see [2]). Although the absolute value of the HF energy is usually much larger than the correlation energy, the correlation energy is very important for energy differences. For example, the O ion is not stable at the HF level, and correlations are necessary in order to obtain even qualitative agreement with the experimental result for the electron affinity of oxygen. In solid state physics, NiO is a well known example of a system which is insulating due to correlations. The most widely used method to include correlations in solids is density functional theory (DFT) [3]. DFT has also recently become quite popular for a computationally efficient treatment of exchange and correlation in molecules. However, a systematic improvement towards the exact results is currently not possible with DFT. Wave-function-based methods are more suitable for this purpose. In the last years, Quantum Monte-Carlo calculations have been performed for several systems [4]. Correlations are included here by multiplying the HF wavefunction with a Jastrow factor. An approach more closely related to quantum chemistry is the Local Ansatz [5, 2] where judiciously chosen local excitation operators are applied to HF wavefunctions from CRYS- TAL calculations. Some years ago, an incremental scheme has been proposed and applied in calculations for semiconductors [6, 7], for graphite [8] and for the valence band of diamond [9]; here information on the effect of local excitations on solid-state properties is drawn from calculations using standard quantum chemical program packages. In a recent paper [10] we showed that this method can be successfully extended to ionic solids; we reported results for the correlation contribution to the cohesive energy of MgO. In the present article, we apply the scheme to the cohesive energy of CaO, as a second example. In addition, we show how correlations affect the lattice constants of MgO and CaO. For these systems, several calculations have been performed at the HF level with the CRYSTAL code [11, 12, 13, 14, 15, 16] as well as with inclusion of correlations using DFT [12, 14, 15, 17]. 1
3 2 The Method The method of increments can be used to build up correlation effects in solids from local correlation contributions which in term may be obtained by transferring results from suitably embedded finite clusters to the infinite crystal. It has been fully described in [6, 7, 8, 10], and a formal derivation has been given within the framework of the projection technique [18]. Thus, we will only briefly repeat the main ideas. (a) Starting from self-consistent field (SCF) calculations localized orbitals are generated which are assumed to be similar in the clusters and in the solid. (b) One-body correlation-energy increments are calculated: in our specific case these are the correlation energies ǫ(a), ǫ(b), ǫ(c),... of localized orbital groups which can be attributed to X 2+ (X = Mg, Ca) or O 2 ions at ionic positions A, B, C,... Each localized orbital group is correlated separately. (c) Two-body increments are defined as non-additivity corrections: ǫ(ab) = ǫ(ab) ǫ(a) ǫ(b), where ǫ(ab) is the correlation energy of the joint orbital system of AB. (d) Three-body increments are defined as ǫ(abc) = ǫ(abc) [ǫ(a)+ǫ(b)+ǫ(c)] [ ǫ(ab)+ ǫ(ac)+ ǫ(bc)]. Similar definitions apply to higher-body increments. (e) The correlation energy of the solid can now be expressed as the sum of all possible increments: ǫ bulk = A ǫ(a)+ 1 ǫ(ab)+ 1 ǫ(abc)+... (1) 2 3! A,B A,B,C Of course, this only makes sense if the incremental expansion is well convergent, i.e. if ǫ(ab) rapidly decreases with increasing distance of the ions at position A and B and if the three-body terms are significantly smaller than the two-body ones. A pre-requisite is that the correlation method used for evaluating the increments must be size-extensive: otherwise the two-body increment ǫ(ab) for two ions A and B at infinite distance would not vanish. In our present work, we used three different size-extensive approaches, cf. Sect Finally, the increments must be transferable, i.e. they should only weakly depend on the cluster chosen. 2
4 2.1 Correlation Methods In this section we want to give a brief description of the correlation methods used. In the averaged coupled-pair functional (ACPF [19]) scheme, the correlation energy is expressed in the form E corr [Ψ c ] = < Ψ SCF +Ψ c H E SCF Ψ SCF +Ψ c > 1+g c < Ψ c Ψ c > (2) withψ SCF beingthescf-wavefunction(usuallyofthespin-restrictedhartree- Fock type) and Ψ c the correlation part of the wavefunction, Ψ c >= a r c r a a+ r a a Ψ SCF > + a<b r<s c rs ab a+ r a+ s a aa b Ψ SCF >; (3) g c is chosen as 2 n in order to make the expression (2) approximately sizeconsistent (n being the number of correlated electrons). For more details (and the extension to multi-reference cases), see [19]. In the coupled-cluster singles and doubles (CCSD [20]) scheme, the wavefunction is expressed with the help of an exponential ansatz: Ψ CCSD >= exp( a r c r a a+ r a a + a<b r<s c rs ab a+ r a+ s a aa b ) Ψ SCF >. (4) a + (a) are creation (annihilation) operators of electrons in orbitals which are occupied (a, b) or unoccupied (r, s) in the SCF wavefunction. Finally, in the CCSD(T) scheme, three particle excitations are included by means of perturbation theory as proposed in [21]. We used these three methods to compare their quality in applications to solids. It turns out that ACPF and CCSD give very similar results, while CCSD(T) yields slighty improved energies [22]. Altogether, the results are not strongly dependent on the methods and no problem arises, therefore, if only one method should be applicable (as is the case for low-spin openshell systems, where CCSD and CCSD(T) are not yet readily available). All calculations of this work were done by using the program package MOLPRO [23, 24]. 3 Cohesive Energy of CaO 3
5 3.1 Basis sets and test calculations For calculating the correlation contribution to the cohesive energy of CaO, wecloselyfollowtheapproachofref.[10]. Foroxygenwechoosea[5s4p3d2f] basis set[25]. Calcium is described by a small-core pseudopotential replacing the 1s, 2s and 2p electrons [26], and a corresponding [6s6p5d2f 1g] valence basisset(fromref. [26], augmentedwithpolarizationfunctionsf 1 = , f 2 =2.142 andg=1.66) is used. All orbitals arecorrelated, withtheexception of the O 1s core. In particular, the correlation contribution of the outercore Ca 3s and 3p orbitals is explicitly taken into account. We did not use a large-core (X 2+ ) pseudopotential and a core polarization potential (CPP) for treating core-valence correlation as was done in the case of MgO [10], because Ca is close to the transition metals and excitations into d-orbitals are important. The influence of the latter on the X 2+ core cannot be well represented by a CPP since the 3d orbitals are core-like themselves (cf. the discussion in [27]). Correlating the Ca outer-core orbitals explicitly, using the small-core (Ca 10+ ) pseudopotential, we circumvent this problem. Using this approach, we performed test calculations for the first and second ionization potential of the Ca atom (Table 1) and calculated spectroscopic properties of the CaO molecule (Table 2). In both cases, we obtain good agreement with experiment. 3.2 Intra-atomic Correlation We first calculated one-body correlation-energy increments. For Ca 2+, the results are virtually independent of the solid-state surroundings. This was tested by doing calculations for a free Ca 2+ and a Ca 2+ embedded in point charges. (A cube of 7x7x7 ions was simulated by point charges ±2, with charges at the surface planes/edges/corners reduced by factors 2/4/8, respectively.) In the case of O 2, of course, the solid-state influence is decisive for stability, and we took it into account by using an embedding similar to that of Ref. [10]: the Pauli repulsion of the 6 nearest Ca 2+ neighbours was simulated by large-core pseudopotentials [28], while the rest of a cube of 7x7x7 lattice sites was treated in point-charge approximation again. A NaCl-like structure with a lattice constant of 4.81 Å was adopted. (The experimental value for the lattice constant is Å at a temperature of T=17.9 K [29]). We performed similar calculations for various other finitecluster approximations of the CaO crystal, in order to insure that the results 4
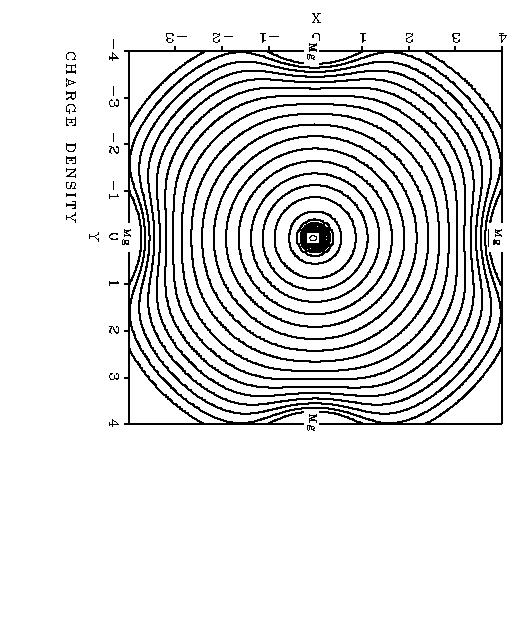
6 are not sensitive to lattice extensions beyond the cube mentioned above. The results for the one-body correlation-energy increments are shown in Table 3. It is interesting to note that the absolute value of the Ca Ca 2+ increment is larger than in the case of Mg, although the electron density in the valence region of Ca is lower than for Mg. The larger correlation contribution for Ca can be rationalized by the fact that excitations into low-lying unoccupied d-orbitals are much more important for Ca than for Mg. This is a result which would be difficult to explain by density functional theory: in a local-density framework, higher density leads to a higher absolute value of the correlation energy. In Ref.[10] we argued that the increment in correlation energy ǫ(embedded O 2 ) ǫ(free O) is not just twice the increment ǫ(free O ) ǫ(free O). However, comparing the increments ǫ (embedded O 2 ) ǫ(embedded O) and ǫ ( embedded O ) ǫ(embedded O) one finds a factor very close to two. This can be seen from Table 4 where we compare the increments in the case of MgO. Thus, for the embedded species linear scaling is appropriate as in the case of the gas-phase isoelectronic series Ne, Ne +, Ne 2+ : there, the increments in correlation energy are H (Ne 2+ Ne + ) and H (Ne + Ne) [30]. Table 4 also shows that the correlation contribution to the electron affinity of the oxygen atom is smaller for the embedded species than in the gas phase. This is dueto the fact that energy differences to excited-state configurations becomelarger whenenclosing O n in asolid-state cage. Once again, this is at variance with a LDA description as the electron density in the case of the embedded O is more compressed than in the case of a free O. In Figure 1 we show the charge density distribution of O 2, again in the case of MgO. We used basis functions on both O and Mg; the Mg 1s, 2s and 2p-electrons are replaced by a pseudopotential. One recognizes the minimum near the Mg 2+ cores, where the Pauli repulsion prevents the oxygen electrons from penetrating into the Mg 2+ core region. This way, the solid is stabilized. The 6 th contour line, counting from Mg to O, is the line which represents a density of a.u. This is the density which encloses about 95 % of the charge and was proposed as an estimate of the size of atoms and molecules [31]. The sum of the intra-ionic correlation-energy increments discussed in this subsection turns out to yield only 60 % of the correlation contribution of the cohesive energy of CaO. This percentage is quite similar to that obtained for MgO [10], at the same level. Thus, although MgO and CaO are to a very good approximation purely ionic solids, the inter-atomic correlation effects 5
7 to be dealt with in the next subsection play an important role. 3.3 Two- and three-body increments When calculating two-body correlation-energy increments, point charges or pseudopotentials surrounding a given ion have to be replaced by real ions. In the case of an additional real O 2, its next-neighbour shell also has to be replaced by a cage of pseudopotentials simulating Ca 2+. This way the increments shown in Table 3 are obtained. Itturnsout that theca-o increments aremuchmoreimportant thanthe O-O increments, while Ca-Ca increments are negligibly small. The changes with respect to MgO [10] can easily be rationalized: On the one hand, the lattice constant is larger than in the case of MgO (4.81 Å vs Å), which reduces the van der Waals interaction and makes the O-O increments smaller. On the other hand, the polarizability of Ca 2+ is by a factor of more than 6 higher than that of Mg 2+ (see for example Ref. [28]), which leads to large Ca-O increments. We show the van der Waals-like decay in Figure 2 by plotting the two-body increments O-O for CaO from CCSD calculations (without including weight factors). By multiplying with the sixth power of the distance, one can verify the van der Waals-law. Plots for the other two-body increments are qualitatively similar. Three-body increments contribute with less than 2 % to the correlation piece of the bulk cohesive energy and may safely be neglected, therefore. A survey of the convergency pattern of the incremental expansion, for both CaO and MgO, is given in Figs. 3 and Sum of increments Adding up the increments of sections 3.2 and 3.3 (cf. Table 5), we obtain between 71 and 78% of the experimental correlation contribution to the cohesive energy which we define as the difference of the experimental cohesive energy (11.0 ev, [32]) plus the zero-point energy (which is taken into account within the Debye approximation and is of the order 0.1 ev) minus the HF binding energy (7.6 ev, Ref. [13]). The percentage obtained is slightly less compared to the case of MgO [10] where 79 to 86 % were recovered. One of the reasons for this difference is that we used a CPP in the case of MgO which covers nearly 100% of the core-valence correlation contributions in Mg, while the explicit treatment of that correlation piece for Ca was less exhaustive. Another reason is that on the Hartree-Fock level f-functions for 6
8 Ca (which are not yet implemented in CRYSTAL) would probably increase the cohesive energy and lower the experimental correlation contribution. Finally, as in the case of MgO, a significant part of the missing correlation energy should be dueto basis set errors for the O atom. Thetotal cohesive energy recovered in our calculations is in the range of between 91 and 93 % of the experimental value. Our results are compared in Table 5 to those from density functional calculations. We choose the results from [12] where a correlation-only functional was used and to H of the correlation contribution to the cohesive energy were obtained, depending on the specific correlation functional used. 4 Lattice constants At the Hartree-Fock level, the lattice constant is in good agreement with experiment for MgO [11, 12, 14, 15, 16], whereas there is a deviation of 0.05 Å in the case of CaO [13]. It is interesting, therefore, to study the influence of correlation effects on lattice constants. In Tables 6 and 7, we give the necessary increments for MgO and CaO, respectively. We find two main effects of correlations. On the one hand, the van der Waals-interaction leads to a reduction of the lattice spacing since the attractive interaction is of the form 1 r and obviously stronger at shorter distance. On the other hand, we 6 find that the intra-ionic correlation of the O 2 -ion forces a larger constant. This can be understood from the argument that excited configurations are lower in energy and mix more strongly with the ground-state determinant if the O 2 is less compressed as explained in section 3.2. Adding up all these contributions (cf. Table 8), they are found to nearly cancel in the case of MgO and to lead to a reduction of only 0.01 Å. For obtaining this result, we applied a linear fit to the correlation energy and superimposed it on the HF potential curve of Refs. [16, 13]. We checked the validity of the linear approximation by calculating selected increments at other lattice constants. In the case of CaO, the van der Waals-interaction is more important and the lattice constant is reduced to 4.81 Å which is in nice agreement with the experimental value. The lattice constants seem to be in better agreement with the experimental values than those calculated from density functional theory for MgO [14, 15] and CaO [14], where deviations of ± 2 % are found. This is similar to earlier findings for semiconductors [7]. 7
9 5 Conclusion We determined the correlation contribution to the cohesive energy of CaO using an expansion into local increments recently applied to MgO. Making use of quantum-chemical ab-initio configuration-interaction calculations for evaluating individual increments, we obtain 80 % of the expected value. The missing energy is probably mainly due to the lack of g and higher polarization functions in our one-particle basis set. The computed lattice constants show deviations of less than 1% from the experimental values. We found two correlation effects on the lattice constants: the inter-atomic van der Waals-force leads to a reduction, whereas intra-atomic correlations of the O 2 ions lead to an increase of the lattice constant. The main difference between CaO and MgO is the reduced importance of the inter-atomic O-O correlations in CaO (due to the larger lattice constant) and the higher importance of the Ca-O correlations (due to the higher polarizability of Ca 2+ ). Compared to DFT, the numerical effort of our scheme is significantly higher. However, we feel that the advantage of the present approach is the high quality and stability of the results both for atoms, ions as well as for solids. Another advantage is the possibility of a systematic improvement by using larger basis sets. We think that the method of local increments is capable now of being routinely applied to ionic systems, and a systematic study on alkali halides is underway. An extension to open-shell systems such as NiO is also a project currently under investigation. Acknowledgments We would like to thank Prof. P. Fulde for supporting this work and Prof. W. C. Nieuwpoort (Groningen) for interesting suggestions. We are grateful to Prof. H.-J. Werner(Stuttgart) for providing the program package MOLPRO. References [1] R. Dovesi, C. Pisani, and C. Roetti, Int. J. Quantum Chem. 17, 517 (1980); C. Pisani, R. Dovesi, and C. Roetti, Lecture Notes in Chemistry, vol. 48 (Springer, Berlin, 1988) 8
10 [2] P. Fulde in Electron Correlations in Molecules and Solids, Springer Series in Solid State Sciences, vol. 100 (Springer, Berlin, 1993) [3] For a review about DFT, see, e.g. R. O. Jones, O. Gunnarsson, Rev. Mod. Phys. 61, 689 (1989) [4] S. Fahy, X. W. Wang, S. G. Louie, Phys. Rev. Lett. 61, 1631 (1988); S. Fahy, X. W. Wang, S. G. Louie, Phys. Rev. B 42, 3503 (1990); X.-P. Li, D. M. Ceperley, R. M. Martin, Phys. Rev. B 44, (1991); L. Mitá s, R. M. Martin, Phys. Rev. Lett. 72, 2438 (1994) [5] G. Stollhoff, P. Fulde, J. Chem. Phys. 73, 4548 (1980) [6] H. Stoll, Phys. Rev. B 46, 6700 (1992); H. Stoll, Chem. Phys. Lett. 191, 548 (1992) [7] B. Paulus, P. Fulde, H. Stoll, Phys. Rev. B 51, (1995) [8] H. Stoll, J. Chem. Phys. 97, 8449 (1992) [9] J. Gräfenstein, H. Stoll, P. Fulde, Chem. Phys. Lett. 215, 611 (1993) [10] K. Doll, M. Dolg, P. Fulde, H. Stoll, Phys. Rev. B 52, 4842 (1995) [11] M. Causà, R.Dovesi, C.Pisani, C.Roetti, Phys.Rev.B33, 1308 (1986) [12] R. Dovesi, C. Roetti, C. Freyria-Fava, E. Aprà, V.R. Saunders, and N.M. Harrison, Philos. Trans. R. Soc. London Ser. A 341, 203 (1992); [13] W. C. Mackrodt, N. M. Harrison, V. R. Saunders, N. L. Allan, M. D. Towler, E. Aprà, R. Dovesi, Philos. Mag. A 68, 653 (1993) [14] M. Causà, A. Zupan, Chem. Phys. Lett. 220, 145 (1994) [15] M. I. McCarthy, N. M. Harrison, Phys. Rev. B 49, 8574 (1994) [16] M. Catti, G. Valerio, R. Dovesi, M. Causà, Phys. Rev. B 49, (1994) [17] N. C. Pyper, Phil. Trans. R. Soc. London A 352, 89 (1995) [18] T. Schork, Thesis, University Stuttgart (1992) [19] R. J. Gdanitz, R. Ahlrichs, Chem. Phys. Lett. 143, 413 (1988) 9
11 [20] F. Koester, H. Kümmel, Nucl. Phys. 17, 477 (1960); J. Číček, J. Chem. Phys. 45, 4256 (1966); J. Paldus, J. Číček, Phys. Rev. A 5, 50 (1972); R. F. Bishop, Theor. Chim. Acta 80, 95 (1991); G. D. Purvis III, R. J. Bartlett, J. Chem. Phys. 76, 1910 (1982) [21] K. Raghavachari, G. W. Trucks, J. A. Pople, M. Head-Gordon, Chem. Phys. Lett. 157, 479 (1989) [22] It was pointed out by H.-J. Werner that CCSD(T) as implemented in Ref. 23 can not be applied to localized orbitals but only to canonical ones (see M. J. O. Deegan, P. J. Knowles, Chem. Phys. Lett. 227, 321 (1994)). In our scheme localization is necessary, but we can estimate the magnitude of the concomitant errors by calculating two-body increments using different clusters which makes localization unnecessary: a cluster with two oxygen atoms, e.g., in order to get ǫ(ab) and a cluster with one oxygen atom to get ǫ(a). We compared the increments ǫ(ab) calculated by using localized/canonical orbitals and found that the error for the most important increment is less than 0.01 ev including the weight factor. The ACPF and CCSD results are not affected from this problem. [23] MOLPRO is a package of ab initio programs written by H.-J. Werner and P.J. Knowles, with contributions from J. Almlöf, R.D. Amos, M.J.O. Deegan, S.T. Elbert, C. Hampel, W. Meyer, K. Peterson, R. Pitzer, A.J. Stone, and P.R. Taylor; the CPP program was written by A. Nicklass. [24] H.-J. Werner and P.J. Knowles, J. Chem. Phys. 82, 5053 (1985); P.J. Knowles and H.-J. Werner, Chem. Phys. Lett. 115, 259 (1985); H.-J. Werner and P.J. Knowles, J. Chem. Phys. 89, 5803(1988); P.J. Knowles and H.-J. Werner, Chem. Phys. Lett. 145, 514 (1988); H.-J. Werner and P.J. Knowles, Theor. Chim. Acta 78, 175 (1990); C. Hampel, K. Peterson, and H.-J. Werner, Chem. Phys. Lett. 190, 1 (1992); P.J. Knowles, C. Hampel, and H.-J. Werner, J. Chem. Phys. 99, 5219 (1993) [25] T.H. Dunning, Jr., J. Chem. Phys. 90, 1007 (1989) [26] M. Kaupp, P. v. R. Schleyer, H. Stoll, H. Preuss, J. Chem. Phys. 94, 1360(1991) 10
12 [27] W. Müller, J. Flesch, and W. Meyer, J. Chem. Phys. 80, 3297 (1984) [28] P. Fuentealba, L. v. Szentpály, H. Preuss and H. Stoll, J. Phys. B 18, 1287 (1985) [29] K.-H. Hellwege and A. M. Hellwege (eds.), Landolt-Börnstein tables, Group III, Vol. 7b1 (Springer, Berlin, 1975) [30] E.R. Davidson, S.A. Hagstrom, S.J. Chakravorty, V.M. Umar, and Ch. Froese-Fischer, Phys. Rev. A 44, 7071 (1991) [31] R. F. Bader, W. H. Henneker, P. E. Cade, J. Chem. Phys. 46, 3341 (1967) [32] CRC Handbook of Chemistry ad Physics, 75 th edition, Editor: David R. Lide (CRC Press, Boca Raton, 1994/1995) [33] C. E. Moore, Atomic Energy Levels, NSRDS-NBS 35 / Vol. I-III, Nat. Bur. Standards (Washington, DC) [34] K. P. Huber, G. Herzberg, Molecular Spectra and Molecular Structure: IV. Constants of Diatomic Molecules (Van Nostrand, New York, 1979) [35] J. A. Irvin, P. J. Dagdigian, J. Chem. Phys. 73, 176 (1980) 11
13 Table 1: Atomic ionization potentials Ca Ca + /Ca + Ca 2+ (in ev) RHF 5.16/11.35 ACPF 6.01/11.78 CCSD 6.03/11.77 CCSD(T) 6.09/11.80 expt. [33] 6.11/11.87 Table 2: Bond length R e (Å), dissociation energy D e (ev) and vibrational frequency ω e (cm 1 ) of the CaO molecule. R e D e ω e RHF CCSD CCSD(T) expt. [34, 35] ±
14 Table 3: Local increments (a.u.) for CaO at a lattice constant of 4.81 Å. weight a ACPF CCSD CCSD(T) Ca Ca O O sum of one-body increments Ca O next neighbour Ca O, 2 nd next neighbour Ca O, 3 rd next neighbour Ca O, 4 th next neighbour Ca Ca, next neighbour Ca Ca, 2 nd next neighbour Ca Ca, 3 rd next neighbour O O, next neighbour O O, 2 nd next neighbour O O, 3 rd next neighbour O O, 4 th next neighbour O O, 5 th next neighbour O O, 6 th next neighbour sum of two-body-increments O O O b O O O c O Ca Ca d O Ca Ca e O Ca O f O Ca O g O Ca O h sum of three-body increments total sum a Weight factor in the incremental expansion of the bulk correlation energy (in a.u. per primitive unit cell) of CaO. b ions at (1,0,0), (0,1,0) and (0,0,1) c ions at (1,0,0), (-1,0,0) and (0,0,1) d O at (0,0,0), Ca at (0,0,1) and (0,1,0) e O at (0,0,0), Ca at (0,0,1) and (0,0,-1) f O at (0,0,0) and (0,1,1), Ca at (0,1,0) g O at (1,0,0) and (-1,0,0), Ca at (0,0,0) h O at (0,0,0) and (0,1,1), Ca at (1,0,0) 13
15 Table 4: Intra-ionic correlation of free and embedded oxygen (in a.u.). incr. O O incr. O O 2 O and O free, O 2 embedded O, O, O 2 embedded Table 5: Correlation contributions to the cohesive energy of CaO (in a.u.). ACPF CCSD CCSD(T) DFT expt [12] Table 6: Local increments (in a.u.) for MgO at a lattice constant of 4.18 Å ACPF CCSD CCSD(T) Mg Mg O O one-body increments Mg-O increments O-O increments two-body increments three-body increments sum at 4.18 Å sum at 4.21 Å
16 Table 7: Local increments (in a.u.) for CaO at a lattice constant of Å ACPF CCSD CCSD(T) Ca Ca O O one-body increments Ca-O increments O-O increments Ca-Ca increments two-body increments three-body increments sum at Å sum at 4.81 Å Table 8: Lattice constants of MgO and CaO (in Å). System RHF ACPF CCSD CCSD(T) DFT exp. MgO [16] [14] [29] [15] CaO [13] [14] [29] 15
17 Figure 1: Charge density of embedded O 2 Figure 2: Van der Waals-like decay of the two-body O-O increments in CaO Figure 3: Sum of local increments for MgO Figure 4: Sum of local increments for CaO 16
18
19 Two body increments: oxygen oxygen CCSD Increment [mh] Increment [mev] nearest neighbour 0.0
20 MgO: sum of increments CCSD [Hartree] one body increments two body increments three body increments sum experimental value [ev] O O Mg O
21 CaO: sum of increments CCSD O O Ca O [Hartree] one body increments two body increments three body increments sum experimental value [ev]
Correlation effects in MgO and CaO: Cohesive energies and lattice constants
 PHYSICAL REVIEW B VOLUME 54, NUMBER 19 15 NOVEMBER 1996-I Correlation effects in MgO and CaO: Cohesive energies and lattice constants Klaus Doll and Michael Dolg Max-Planck-Institut für Physik Komplexer
PHYSICAL REVIEW B VOLUME 54, NUMBER 19 15 NOVEMBER 1996-I Correlation effects in MgO and CaO: Cohesive energies and lattice constants Klaus Doll and Michael Dolg Max-Planck-Institut für Physik Komplexer
arxiv:cond-mat/ v1 10 May 1996
 Cohesive energies of cubic III-V semiconductors Beate Paulus, Peter Fulde Max-Planck-Institut für Physik komplexer Systeme, Bayreuther Str. 40, 01187 Dresden, Germany arxiv:cond-mat/9605064v1 10 May 1996
Cohesive energies of cubic III-V semiconductors Beate Paulus, Peter Fulde Max-Planck-Institut für Physik komplexer Systeme, Bayreuther Str. 40, 01187 Dresden, Germany arxiv:cond-mat/9605064v1 10 May 1996
Ab initio treatment of electron correlations in polymers: Lithium hydride
 JOURNAL OF CHEMICAL PHYSICS VOLUME 112, NUMBER 10 8 MARCH 2000 Ab initio treatment of electron correlations in polymers: Lithium hydride chain and beryllium hydride polymer Ayjamal Abdurahman a) Max-Planck-Institut
JOURNAL OF CHEMICAL PHYSICS VOLUME 112, NUMBER 10 8 MARCH 2000 Ab initio treatment of electron correlations in polymers: Lithium hydride chain and beryllium hydride polymer Ayjamal Abdurahman a) Max-Planck-Institut
Quantum-chemical approach to cohesive properties of metallic beryllium
 Quantum-chemical approach to cohesive properties of metallic beryllium Elena Voloshina 1, Beate Paulus 2, and Hermann Stoll 3 1 Max-Planck-Institut für Physik komplexer Systeme, Nöthnitzer Straße 38, 01187
Quantum-chemical approach to cohesive properties of metallic beryllium Elena Voloshina 1, Beate Paulus 2, and Hermann Stoll 3 1 Max-Planck-Institut für Physik komplexer Systeme, Nöthnitzer Straße 38, 01187
Correlated ab initio calculations for ground-state properties of II-VI semiconductors
 PHYSICAL REVIEW B VOLUME 56, NUMBER 12 15 SEPTEMBER 1997-II Correlated ab initio calculations for ground-state properties of II-VI semiconductors Martin Albrecht and Beate Paulus Max-Planck-Institut für
PHYSICAL REVIEW B VOLUME 56, NUMBER 12 15 SEPTEMBER 1997-II Correlated ab initio calculations for ground-state properties of II-VI semiconductors Martin Albrecht and Beate Paulus Max-Planck-Institut für
Wavefunction-based Correlation Calculations for Hole and Electron Capture States in Solids and Polymers
 Wavefunction-based orrelation alculations for ole and Electron apture States in Solids and Polymers Uwe Birkenheuer 1, hrista Willnauer, Malte von Arnim, Walter Alsheimer, Dmitry Izotov Wavefunction-based
Wavefunction-based orrelation alculations for ole and Electron apture States in Solids and Polymers Uwe Birkenheuer 1, hrista Willnauer, Malte von Arnim, Walter Alsheimer, Dmitry Izotov Wavefunction-based
ELECTRONIC STRUCTURE OF MAGNESIUM OXIDE
 Int. J. Chem. Sci.: 8(3), 2010, 1749-1756 ELECTRONIC STRUCTURE OF MAGNESIUM OXIDE P. N. PIYUSH and KANCHAN LATA * Department of Chemistry, B. N. M. V. College, Sahugarh, MADHIPUR (Bihar) INDIA ABSTRACT
Int. J. Chem. Sci.: 8(3), 2010, 1749-1756 ELECTRONIC STRUCTURE OF MAGNESIUM OXIDE P. N. PIYUSH and KANCHAN LATA * Department of Chemistry, B. N. M. V. College, Sahugarh, MADHIPUR (Bihar) INDIA ABSTRACT
Valence-band structure of group-iv semiconductors by means of local increments
 PHYSICAL REVIEW B VOLUME 55, NUMBER 20 15 MAY 1997-II Valence-band structure of group-iv semiconductors by means of local increments Jürgen Gräfenstein* Max-Planck-Institut für Physik komplexer Systeme
PHYSICAL REVIEW B VOLUME 55, NUMBER 20 15 MAY 1997-II Valence-band structure of group-iv semiconductors by means of local increments Jürgen Gräfenstein* Max-Planck-Institut für Physik komplexer Systeme
The lattice structure of mercury: Influence of electronic correlation
 The lattice structure of mercury: Influence of electronic correlation Nicola Gaston, Beate Paulus, and Krzysztof Rosciszewski Max-Planck-Institut für Physik komplexer Systeme, Nöthnitzer Straße 38, D-01187
The lattice structure of mercury: Influence of electronic correlation Nicola Gaston, Beate Paulus, and Krzysztof Rosciszewski Max-Planck-Institut für Physik komplexer Systeme, Nöthnitzer Straße 38, D-01187
Calculations of band structures
 Chemistry and Physics at Albany Planning for the Future Calculations of band structures using wave-function based correlation methods Elke Pahl Centre of Theoretical Chemistry and Physics Institute of
Chemistry and Physics at Albany Planning for the Future Calculations of band structures using wave-function based correlation methods Elke Pahl Centre of Theoretical Chemistry and Physics Institute of
Cohesive properties of CeN and LaN from first principles
 APS/23-QED Cohesive properties of CeN and LaN from first principles Elena Voloshina a and Beate Paulus b a Max-Planck-Institut für Physik komplexer Systeme, Nöthnitzer Straße 38, 087 Dresden, Germany b
APS/23-QED Cohesive properties of CeN and LaN from first principles Elena Voloshina a and Beate Paulus b a Max-Planck-Institut für Physik komplexer Systeme, Nöthnitzer Straße 38, 087 Dresden, Germany b
The performance of the Hartree-Fock-Wigner correlation model for light diatomic molecules
 The performance of the Hartree-Fock-Wigner correlation model for light diatomic molecules Rebecca Fondermann, Michael Hanrath, Michael Dolg Institut für Theoretische Chemie, Universität zu Köln, Greinstr.
The performance of the Hartree-Fock-Wigner correlation model for light diatomic molecules Rebecca Fondermann, Michael Hanrath, Michael Dolg Institut für Theoretische Chemie, Universität zu Köln, Greinstr.
Ab initio calculations on the ground and low-lying excited states of InI
 MOLECULAR PHYSICS, 1OCTOBER 23, VOL. 11, NO. 19, 2963 2968 Ab initio calculations on the ground and low-lying excited states of InI WENLI ZOU, MEIRONG LIN*, XINZHENG YANG and BAOZHENG ZHANG Institute of
MOLECULAR PHYSICS, 1OCTOBER 23, VOL. 11, NO. 19, 2963 2968 Ab initio calculations on the ground and low-lying excited states of InI WENLI ZOU, MEIRONG LIN*, XINZHENG YANG and BAOZHENG ZHANG Institute of
Introduction to Computational Chemistry
 Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
7/29/2014. Electronic Structure. Electrons in Momentum Space. Electron Density Matrices FKF FKF. Ulrich Wedig
 Electron Density Matrices Density matrices Γ, an alternative to the wavefunction Ψ, for the description of a quantum system Electronic Structure The N-particle density matrix Electrons in Momentum Space
Electron Density Matrices Density matrices Γ, an alternative to the wavefunction Ψ, for the description of a quantum system Electronic Structure The N-particle density matrix Electrons in Momentum Space
PBS: FROM SOLIDS TO CLUSTERS
 PBS: FROM SOLIDS TO CLUSTERS E. HOFFMANN AND P. ENTEL Theoretische Tieftemperaturphysik Gerhard-Mercator-Universität Duisburg, Lotharstraße 1 47048 Duisburg, Germany Semiconducting nanocrystallites like
PBS: FROM SOLIDS TO CLUSTERS E. HOFFMANN AND P. ENTEL Theoretische Tieftemperaturphysik Gerhard-Mercator-Universität Duisburg, Lotharstraße 1 47048 Duisburg, Germany Semiconducting nanocrystallites like
Theoretical determination of the heat of formation of methylene
 Theoretical determination of the heat of formation of methylene Nikos L. Doltsinis and Peter J. Knowles School of Chemistry, University of Birmingham, Edgbaston, Birmingham B5 2TT, United Kingdom The heat
Theoretical determination of the heat of formation of methylene Nikos L. Doltsinis and Peter J. Knowles School of Chemistry, University of Birmingham, Edgbaston, Birmingham B5 2TT, United Kingdom The heat
The high-pressure phase transitions of silicon and gallium nitride: a comparative study of Hartree Fock and density functional calculations
 J. Phys.: Condens. Matter 8 (1996) 3993 4000. Printed in the UK The high-pressure phase transitions of silicon and gallium nitride: a comparative study of Hartree Fock and density functional calculations
J. Phys.: Condens. Matter 8 (1996) 3993 4000. Printed in the UK The high-pressure phase transitions of silicon and gallium nitride: a comparative study of Hartree Fock and density functional calculations
Solution of the Electronic Schrödinger Equation. Using Basis Sets to Solve the Electronic Schrödinger Equation with Electron Correlation
 Solution of the Electronic Schrödinger Equation Using Basis Sets to Solve the Electronic Schrödinger Equation with Electron Correlation Errors in HF Predictions: Binding Energies D e (kcal/mol) HF Expt
Solution of the Electronic Schrödinger Equation Using Basis Sets to Solve the Electronic Schrödinger Equation with Electron Correlation Errors in HF Predictions: Binding Energies D e (kcal/mol) HF Expt
Benchmark calculations with correlated molecular wave functions
 Theor Chem Acc (1997) 97:251±259 Benchmark calculations with correlated molecular wave functions XII. Core correlation e ects on the homonuclear diatomic molecules B 2 -F 2 Kirk A. Peterson 1, Angela K.
Theor Chem Acc (1997) 97:251±259 Benchmark calculations with correlated molecular wave functions XII. Core correlation e ects on the homonuclear diatomic molecules B 2 -F 2 Kirk A. Peterson 1, Angela K.
No. 2 lectronic state and potential energy function for UH where ρ = r r e, r being the interatomic distance and r e its equilibrium value. How
 Vol 12 No 2, February 2003 cfl 2003 Chin. Phys. Soc. 1009-1963/2003/12(02)/0154-05 Chinese Physics and IOP Publishing Ltd lectronic state and potential energy function for UH 2+* Wang Hong-Yan( Ψ) a)y,
Vol 12 No 2, February 2003 cfl 2003 Chin. Phys. Soc. 1009-1963/2003/12(02)/0154-05 Chinese Physics and IOP Publishing Ltd lectronic state and potential energy function for UH 2+* Wang Hong-Yan( Ψ) a)y,
Ab initio coupled-cluster calculations for the fcc and hcp structures of rare-gas solids
 PHYSICAL REVIEW B VOLUME 62, NUMBER 9 1 SEPTEMBER 2000-I Ab initio coupled-cluster calculations for the fcc and hcp structures of rare-gas solids Krzysztof Rościszewski Institute of Physics, Jagellonian
PHYSICAL REVIEW B VOLUME 62, NUMBER 9 1 SEPTEMBER 2000-I Ab initio coupled-cluster calculations for the fcc and hcp structures of rare-gas solids Krzysztof Rościszewski Institute of Physics, Jagellonian
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory
 Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
AN INTRODUCTION TO QUANTUM CHEMISTRY. Mark S. Gordon Iowa State University
 AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
Bonding in solids The interaction of electrons in neighboring atoms of a solid serves the very important function of holding the crystal together.
 Bonding in solids The interaction of electrons in neighboring atoms of a solid serves the very important function of holding the crystal together. For example Nacl In the Nacl lattice, each Na atom is
Bonding in solids The interaction of electrons in neighboring atoms of a solid serves the very important function of holding the crystal together. For example Nacl In the Nacl lattice, each Na atom is
Basis set convergence in extended systems: infinite hydrogen fluoride and hydrogen chloride chains
 Chemical Physics Letters 398 (2004) 44 49 www.elsevier.com/locate/cplett Basis set convergence in extended systems: infinite hydrogen fluoride and hydrogen chloride chains Christian Buth *, Beate Paulus
Chemical Physics Letters 398 (2004) 44 49 www.elsevier.com/locate/cplett Basis set convergence in extended systems: infinite hydrogen fluoride and hydrogen chloride chains Christian Buth *, Beate Paulus
arxiv:cond-mat/ v2 [cond-mat.other] 21 Nov 2005
![arxiv:cond-mat/ v2 [cond-mat.other] 21 Nov 2005](/thumbs/72/66976991.jpg "arxiv:cond-mat/ v2 [cond-mat.other] 21 Nov 2005") arxiv:cond-mat/0408243v2 [cond-mat.other] 21 Nov 2005 Basis set convergence in extended systems: infinite hydrogen fluoride and hydrogen chloride chains Christian Buth, Beate Paulus Max-Planck-Institut
arxiv:cond-mat/0408243v2 [cond-mat.other] 21 Nov 2005 Basis set convergence in extended systems: infinite hydrogen fluoride and hydrogen chloride chains Christian Buth, Beate Paulus Max-Planck-Institut
Relativistic and correlated calculations on the ground, excited, and ionized states of iodine
 Relativistic and correlated calculations on the ground, excited, and ionized states of iodine W. A. de Jong, L. Visscher, a) and W. C. Nieuwpoort Laboratory for Chemical Physics and Materials Science Centre,
Relativistic and correlated calculations on the ground, excited, and ionized states of iodine W. A. de Jong, L. Visscher, a) and W. C. Nieuwpoort Laboratory for Chemical Physics and Materials Science Centre,
3: Many electrons. Orbital symmetries. l =2 1. m l
 3: Many electrons Orbital symmetries Atomic orbitals are labelled according to the principal quantum number, n, and the orbital angular momentum quantum number, l. Electrons in a diatomic molecule experience
3: Many electrons Orbital symmetries Atomic orbitals are labelled according to the principal quantum number, n, and the orbital angular momentum quantum number, l. Electrons in a diatomic molecule experience
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Detlev Figgen, Erich Goll, and Hermann Stoll b) Institut für Theoretische Chemie, Universität Stuttgart, D Stuttgart, Germany
 Institut für Theoretische Chemie, Universität Stuttgart, D Stuttgart, Germany") JOURNAL OF CHEMICAL PHYSICS VOLUME 119, NUMBER 21 1 DECEMBER 2003 Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis
JOURNAL OF CHEMICAL PHYSICS VOLUME 119, NUMBER 21 1 DECEMBER 2003 Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis
Gaussian Basis Sets for Solid-State Calculations
 Gaussian Basis Sets for Solid-State Calculations K. Doll Molpro Quantum Chemistry Software Institute of Theoretical Chemistry, D-70569 Stuttgart, Germany MW-MSSC 2017, Minneapolis, July 10, 2017 Introduction
Gaussian Basis Sets for Solid-State Calculations K. Doll Molpro Quantum Chemistry Software Institute of Theoretical Chemistry, D-70569 Stuttgart, Germany MW-MSSC 2017, Minneapolis, July 10, 2017 Introduction
Approximating the basis set dependence of coupled cluster calculations: Evaluation of perturbation theory approximations for stable molecules
 JOURNAL OF CHEMICAL PHYSICS VOLUME 113, NUMBER 18 8 NOVEMBER 2000 Approximating the basis set dependence of coupled cluster calculations: Evaluation of perturbation theory approximations for stable molecules
JOURNAL OF CHEMICAL PHYSICS VOLUME 113, NUMBER 18 8 NOVEMBER 2000 Approximating the basis set dependence of coupled cluster calculations: Evaluation of perturbation theory approximations for stable molecules
Chapter 3. Crystal Binding
 Chapter 3. Crystal Binding Energy of a crystal and crystal binding Cohesive energy of Molecular crystals Ionic crystals Metallic crystals Elasticity What causes matter to exist in three different forms?
Chapter 3. Crystal Binding Energy of a crystal and crystal binding Cohesive energy of Molecular crystals Ionic crystals Metallic crystals Elasticity What causes matter to exist in three different forms?
Calculation of Molecular Constants for the of the NeH + and KrH + Ions
 Calculation of Molecular Constants for the of the NeH and KrH Ions Ground States P. Rosmus and E.-A. Reinsch Fachbereich Chemie der Universität Frankfurt (Main) Z. Naturforsch. a, 66-7 (98); received July,
Calculation of Molecular Constants for the of the NeH and KrH Ions Ground States P. Rosmus and E.-A. Reinsch Fachbereich Chemie der Universität Frankfurt (Main) Z. Naturforsch. a, 66-7 (98); received July,
Electron Correlation - Methods beyond Hartree-Fock
 Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
Correlation in correlated materials (mostly transition metal oxides) Lucas K. Wagner University of Illinois at Urbana-Champaign
 Lucas K. Wagner University of Illinois at Urbana-Champaign") Correlation in correlated materials (mostly transition metal oxides) Lucas K. Wagner University of Illinois at Urbana-Champaign Understanding of correlated materials is mostly phenomenological FN- DMC
Correlation in correlated materials (mostly transition metal oxides) Lucas K. Wagner University of Illinois at Urbana-Champaign Understanding of correlated materials is mostly phenomenological FN- DMC
Ab initio structure prediction for molecules and solids
 Ab initio structure prediction for molecules and solids Klaus Doll Max-Planck-Institute for Solid State Research Stuttgart Chemnitz, June/July 2010 Contents structure prediction: 1) global search on potential
Ab initio structure prediction for molecules and solids Klaus Doll Max-Planck-Institute for Solid State Research Stuttgart Chemnitz, June/July 2010 Contents structure prediction: 1) global search on potential
Towards gas-phase accuracy for condensed phase problems
 Towards gas-phase accuracy for condensed phase problems Fred Manby Centre for Computational Chemistry, School of Chemistry University of Bristol STC 2006: Quantum Chemistry Methods and Applications Erkner,
Towards gas-phase accuracy for condensed phase problems Fred Manby Centre for Computational Chemistry, School of Chemistry University of Bristol STC 2006: Quantum Chemistry Methods and Applications Erkner,
Chemistry 334 Part 2: Computational Quantum Chemistry
 Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
All-electron quantum Monte Carlo calculations for the noble gas atoms He to Xe
 All-electron quantum Monte Carlo calculations for the noble gas atoms He to Xe A. Ma, N. D. Drummond, M. D. Towler, and R. J. Needs Theory of Condensed Matter Group, Cavendish Laboratory, University of
All-electron quantum Monte Carlo calculations for the noble gas atoms He to Xe A. Ma, N. D. Drummond, M. D. Towler, and R. J. Needs Theory of Condensed Matter Group, Cavendish Laboratory, University of
The Nature of the Interlayer Interaction in Bulk. and Few-Layer Phosphorus
 Supporting Information for: The Nature of the Interlayer Interaction in Bulk and Few-Layer Phosphorus L. Shulenburger, A.D. Baczewski, Z. Zhu, J. Guan, and D. Tománek, Sandia National Laboratories, Albuquerque,
Supporting Information for: The Nature of the Interlayer Interaction in Bulk and Few-Layer Phosphorus L. Shulenburger, A.D. Baczewski, Z. Zhu, J. Guan, and D. Tománek, Sandia National Laboratories, Albuquerque,
Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type
 Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type Zhigang Gu, a Lars Heinke, a,* Christof Wöll a, Tobias Neumann,
Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type Zhigang Gu, a Lars Heinke, a,* Christof Wöll a, Tobias Neumann,
Charge renormalization at the large-d limit for N-electron atoms and weakly bound systems
 Charge renormalization at the large-d limit for N-electron atoms and weakly bound systems S. Kais and R. Bleil Department of Chemistry, Purdue University, West Lafayette, Indiana 47907 Received 25 January
Charge renormalization at the large-d limit for N-electron atoms and weakly bound systems S. Kais and R. Bleil Department of Chemistry, Purdue University, West Lafayette, Indiana 47907 Received 25 January
QMC dissociation energy of the water dimer: Time step errors and backflow calculations
 QMC dissociation energy of the water dimer: Time step errors and backflow calculations Idoia G. de Gurtubay and Richard J. Needs TCM group. Cavendish Laboratory University of Cambridge Idoia G. de Gurtubay.
QMC dissociation energy of the water dimer: Time step errors and backflow calculations Idoia G. de Gurtubay and Richard J. Needs TCM group. Cavendish Laboratory University of Cambridge Idoia G. de Gurtubay.
Lecture 2: Bonding in solids
 Lecture 2: Bonding in solids Electronegativity Van Arkel-Ketalaar Triangles Atomic and ionic radii Band theory of solids Molecules vs. solids Band structures Analysis of chemical bonds in Reciprocal space
Lecture 2: Bonding in solids Electronegativity Van Arkel-Ketalaar Triangles Atomic and ionic radii Band theory of solids Molecules vs. solids Band structures Analysis of chemical bonds in Reciprocal space
Open-shell and Magnetic Systems with CRYSTAL Tools
 Open-shell and Magnetic Systems with CRYSTAL Tools Klaus Doll Molpro Quantum Chemistry Software Institute of Theoretical Chemistry, D-70569 Stuttgart, Germany MSSC 2018, Turin, September 2018 Contents
Open-shell and Magnetic Systems with CRYSTAL Tools Klaus Doll Molpro Quantum Chemistry Software Institute of Theoretical Chemistry, D-70569 Stuttgart, Germany MSSC 2018, Turin, September 2018 Contents
Relativistic and correlation effects on molecular properties. I. The dihalogens F 2,Cl 2,Br 2,I 2, and At 2
 Relativistic and correlation effects on molecular properties. I. The dihalogens F 2,Cl 2,Br 2,I 2, and At 2 L. Visscher Laboratory of Chemical Physics and Material Science Center, University of Groningen,
Relativistic and correlation effects on molecular properties. I. The dihalogens F 2,Cl 2,Br 2,I 2, and At 2 L. Visscher Laboratory of Chemical Physics and Material Science Center, University of Groningen,
Theoretical study of spin-orbit coupling constants for O 2
 JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 16 22 OCTOBER 2001 Theoretical study of spin-orbit coupling constants for O 2 A 2 3Õ2,1Õ2u, v Ä0 17 and a 4 5Õ2,3Õ2,1Õ2,À1Õ2u, v Ä0 25 D. G. Fedorov, M. S.
JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 16 22 OCTOBER 2001 Theoretical study of spin-orbit coupling constants for O 2 A 2 3Õ2,1Õ2u, v Ä0 17 and a 4 5Õ2,3Õ2,1Õ2,À1Õ2u, v Ä0 25 D. G. Fedorov, M. S.
Manuel Díaz-Tinoco and J. V. Ortiz Department of Chemistry and Biochemistry Auburn University Auburn AL Abstract
 JCP Comment on Are polynuclear superhalogens without halogen atoms probable? A high level ab initio case study on triple bridged binuclear anions with cyanide ligands [J. Chem. Phys. 140, 094301 (2014)]
JCP Comment on Are polynuclear superhalogens without halogen atoms probable? A high level ab initio case study on triple bridged binuclear anions with cyanide ligands [J. Chem. Phys. 140, 094301 (2014)]
X-Ray transitions to low lying empty states
 X-Ray Spectra: - continuous part of the spectrum is due to decelerated electrons - the maximum frequency (minimum wavelength) of the photons generated is determined by the maximum kinetic energy of the
X-Ray Spectra: - continuous part of the spectrum is due to decelerated electrons - the maximum frequency (minimum wavelength) of the photons generated is determined by the maximum kinetic energy of the
Homologation of Boronic Esters with Organolithium Compounds: A Computational Assessment of Mechanism
 Homologation of Boronic Esters with Organolithium Compounds: A Computational Assessment of Mechanism Stéphanie Essafi,*,1 Simone Tomasi, 2 Varinder K. Aggarwal, 1 Jeremy Harvey*,1 1 School of Chemistry,
Homologation of Boronic Esters with Organolithium Compounds: A Computational Assessment of Mechanism Stéphanie Essafi,*,1 Simone Tomasi, 2 Varinder K. Aggarwal, 1 Jeremy Harvey*,1 1 School of Chemistry,
Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data January 2012
 2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
2327-3 Joint ICTP-IAEA Workshop on Fusion Plasma Modelling using Atomic and Molecular Data 23-27 January 2012 Qunatum Methods for Plasma-Facing Materials Alain ALLOUCHE Univ.de Provence, Lab.de la Phys.
Relativistic and correlation effects on molecular properties. II. The hydrogen halides HF, HCl, HBr, HI, and HAt
 Relativistic and correlation effects on molecular properties. II. The hydrogen halides HF, HCl, HBr, HI, and HAt L. Visscher Laboratory of Chemical Physics and Materials Science Center, University of Groningen,
Relativistic and correlation effects on molecular properties. II. The hydrogen halides HF, HCl, HBr, HI, and HAt L. Visscher Laboratory of Chemical Physics and Materials Science Center, University of Groningen,
DFT EXERCISES. FELIPE CERVANTES SODI January 2006
 DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
Journal of Theoretical Physics
 1 Journal of Theoretical Physics Founded and Edited by M. Apostol 53 (2000) ISSN 1453-4428 Ionization potential for metallic clusters L. C. Cune and M. Apostol Department of Theoretical Physics, Institute
1 Journal of Theoretical Physics Founded and Edited by M. Apostol 53 (2000) ISSN 1453-4428 Ionization potential for metallic clusters L. C. Cune and M. Apostol Department of Theoretical Physics, Institute
OVERVIEW OF QUANTUM CHEMISTRY METHODS
 OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
Beyond the Hartree-Fock Approximation: Configuration Interaction
 Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
Density Functional Theory. Martin Lüders Daresbury Laboratory
 Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Performance of Hartree-Fock and Correlated Methods
 Chemistry 460 Fall 2017 Dr. Jean M. Standard December 4, 2017 Performance of Hartree-Fock and Correlated Methods Hartree-Fock Methods Hartree-Fock methods generally yield optimized geomtries and molecular
Chemistry 460 Fall 2017 Dr. Jean M. Standard December 4, 2017 Performance of Hartree-Fock and Correlated Methods Hartree-Fock Methods Hartree-Fock methods generally yield optimized geomtries and molecular
Quantum Monte Carlo methods
 Quantum Monte Carlo methods Lubos Mitas North Carolina State University Urbana, August 2006 Lubos_Mitas@ncsu.edu H= 1 2 i i 2 i, I Z I r ii i j 1 r ij E ion ion H r 1, r 2,... =E r 1, r 2,... - ground
Quantum Monte Carlo methods Lubos Mitas North Carolina State University Urbana, August 2006 Lubos_Mitas@ncsu.edu H= 1 2 i i 2 i, I Z I r ii i j 1 r ij E ion ion H r 1, r 2,... =E r 1, r 2,... - ground
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
Electron Correlation Methods
 Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
MO Calculation for a Diatomic Molecule. /4 0 ) i=1 j>i (1/r ij )
 i=1 j>i (1/r ij )") MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
v(r i r j ) = h(r i )+ 1 N
 = h(r i )+ 1 N") Chapter 1 Hartree-Fock Theory 1.1 Formalism For N electrons in an external potential V ext (r), the many-electron Hamiltonian can be written as follows: N H = [ p i i=1 m +V ext(r i )]+ 1 N N v(r i r j
Chapter 1 Hartree-Fock Theory 1.1 Formalism For N electrons in an external potential V ext (r), the many-electron Hamiltonian can be written as follows: N H = [ p i i=1 m +V ext(r i )]+ 1 N N v(r i r j
Ab initio calculations for potential energy surfaces. D. Talbi GRAAL- Montpellier
 Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Energy-adjusted pseudopotentials for the rare earth elements
 Theor Chim Acta (1989) 75:173-194 O Springer-Ver!ag 1989 Energy-adjusted pseudopotentials for the rare earth elements M. Dolg, H. Stoll, A. Savin, and H. Preuss Institut f/it Theoretische Chemie, Universit/it
Theor Chim Acta (1989) 75:173-194 O Springer-Ver!ag 1989 Energy-adjusted pseudopotentials for the rare earth elements M. Dolg, H. Stoll, A. Savin, and H. Preuss Institut f/it Theoretische Chemie, Universit/it
COUPLED-CLUSTER CALCULATIONS OF GROUND AND EXCITED STATES OF NUCLEI
 COUPLED-CLUSTER CALCULATIONS OF GROUND AND EXCITED STATES OF NUCLEI Marta Włoch, a Jeffrey R. Gour, a and Piotr Piecuch a,b a Department of Chemistry,Michigan State University, East Lansing, MI 48824 b
COUPLED-CLUSTER CALCULATIONS OF GROUND AND EXCITED STATES OF NUCLEI Marta Włoch, a Jeffrey R. Gour, a and Piotr Piecuch a,b a Department of Chemistry,Michigan State University, East Lansing, MI 48824 b
Electronic structure of lanthanide dimers
 MOLECULAR PHYSICS, 10 July 2003, VOL. 101, NO. 13, 1967 1976 Electronic structure of lanthanide dimers XIAOYAN CAO 1,2 and MICHAEL DOLG 1, * 1 Institut fu r Theoretische Chemie, Universita tzuko ln, D-50939,
MOLECULAR PHYSICS, 10 July 2003, VOL. 101, NO. 13, 1967 1976 Electronic structure of lanthanide dimers XIAOYAN CAO 1,2 and MICHAEL DOLG 1, * 1 Institut fu r Theoretische Chemie, Universita tzuko ln, D-50939,
The broad topic of physical metallurgy provides a basis that links the structure of materials with their properties, focusing primarily on metals.
 Physical Metallurgy The broad topic of physical metallurgy provides a basis that links the structure of materials with their properties, focusing primarily on metals. Crystal Binding In our discussions
Physical Metallurgy The broad topic of physical metallurgy provides a basis that links the structure of materials with their properties, focusing primarily on metals. Crystal Binding In our discussions
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
MRCI calculations in MOLPRO
 1 MRCI calculations in MOLPRO Molpro is a software package written in Fortran and maintained by H.J. Werner and P.J. Knowles. It is often used for performing sophisticated electronic structure calculations,
1 MRCI calculations in MOLPRO Molpro is a software package written in Fortran and maintained by H.J. Werner and P.J. Knowles. It is often used for performing sophisticated electronic structure calculations,
Periodic Trends in Properties of Homonuclear
 Chapter 8 Periodic Trends in Properties of Homonuclear Diatomic Molecules Up to now, we have discussed various physical properties of nanostructures, namely, two-dimensional - graphene-like structures:
Chapter 8 Periodic Trends in Properties of Homonuclear Diatomic Molecules Up to now, we have discussed various physical properties of nanostructures, namely, two-dimensional - graphene-like structures:
Radiative Transition Probabilities and Lifetimes for the Band Systems A 2 Π X 2 Σ + of the Isovalent Molecules BeF, MgF and CaF
 950 Brazilian Journal of Physics, vol. 35, no. 4A, December, 2005 Radiative Transition Probabilities and Lifetimes for the Band Systems of the Isovalent Molecules BeF, MgF and CaF Marina Pelegrini a, Ciro
950 Brazilian Journal of Physics, vol. 35, no. 4A, December, 2005 Radiative Transition Probabilities and Lifetimes for the Band Systems of the Isovalent Molecules BeF, MgF and CaF Marina Pelegrini a, Ciro
A local MP2 periodic study of crystalline argon
 Journal of Physics: Conference Series A local MP2 periodic study of crystalline argon To cite this article: S Casassa et al 2008 J. Phys.: Conf. Ser. 117 012007 Recent citations - Laplace transformed MP2
Journal of Physics: Conference Series A local MP2 periodic study of crystalline argon To cite this article: S Casassa et al 2008 J. Phys.: Conf. Ser. 117 012007 Recent citations - Laplace transformed MP2
Accurate multireference configuration interaction calculations on the lowest 1 and 3 electronic states of C 2,CN, BN, and BO
 Accurate multireference configuration interaction calculations on the lowest 1 and 3 electronic states of C 2,CN, BN, and BO Kirk A. Peterson a) Department of Chemistry, Washington State University and
Accurate multireference configuration interaction calculations on the lowest 1 and 3 electronic states of C 2,CN, BN, and BO Kirk A. Peterson a) Department of Chemistry, Washington State University and
Assessment of range-separated time-dependent density-functional theory for calculating C 6 dispersion coefficients
 1/10 Assessment of range-separated time-dependent density-functional theory for calculating C 6 dispersion coefficients Julien Toulouse 1,2, Elisa Rebolini 1, Tim Gould 3, John F. Dobson 3, Prasenjit Seal
1/10 Assessment of range-separated time-dependent density-functional theory for calculating C 6 dispersion coefficients Julien Toulouse 1,2, Elisa Rebolini 1, Tim Gould 3, John F. Dobson 3, Prasenjit Seal
Principles of Quantum Mechanics
 Principles of Quantum Mechanics - indistinguishability of particles: bosons & fermions bosons: total wavefunction is symmetric upon interchange of particle coordinates (space,spin) fermions: total wavefuncftion
Principles of Quantum Mechanics - indistinguishability of particles: bosons & fermions bosons: total wavefunction is symmetric upon interchange of particle coordinates (space,spin) fermions: total wavefuncftion
Electronic Supplementary Information
 Electronic Supplementary Material (ESI) for CrystEngComm. This journal is The Royal Society of Chemistry 2014 Electronic Supplementary Information Configurational and energetical study of the (100) and
Electronic Supplementary Material (ESI) for CrystEngComm. This journal is The Royal Society of Chemistry 2014 Electronic Supplementary Information Configurational and energetical study of the (100) and
The Gutzwiller Density Functional Theory
 The Gutzwiller Density Functional Theory Jörg Bünemann, BTU Cottbus I) Introduction 1. Model for an H 2 -molecule 2. Transition metals and their compounds II) Gutzwiller variational theory 1. Gutzwiller
The Gutzwiller Density Functional Theory Jörg Bünemann, BTU Cottbus I) Introduction 1. Model for an H 2 -molecule 2. Transition metals and their compounds II) Gutzwiller variational theory 1. Gutzwiller
Lecture 6 - Bonding in Crystals
 Lecture 6 onding in Crystals inding in Crystals (Kittel Ch. 3) inding of atoms to form crystals A crystal is a repeated array of atoms Why do they form? What are characteristic bonding mechanisms? How
Lecture 6 onding in Crystals inding in Crystals (Kittel Ch. 3) inding of atoms to form crystals A crystal is a repeated array of atoms Why do they form? What are characteristic bonding mechanisms? How
Introduction to Density Functional Theory
 1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
Metal-insulator transitions
 Metal-insulator transitions What can we learn from electronic structure calculations? Mike Towler mdt26@phy.cam.ac.uk www.tcm.phy.cam.ac.uk/ mdt26 Theory of Condensed Matter Group Cavendish Laboratory
Metal-insulator transitions What can we learn from electronic structure calculations? Mike Towler mdt26@phy.cam.ac.uk www.tcm.phy.cam.ac.uk/ mdt26 Theory of Condensed Matter Group Cavendish Laboratory
Section 3 Electronic Configurations, Term Symbols, and States
 Section 3 Electronic Configurations, Term Symbols, and States Introductory Remarks- The Orbital, Configuration, and State Pictures of Electronic Structure One of the goals of quantum chemistry is to allow
Section 3 Electronic Configurations, Term Symbols, and States Introductory Remarks- The Orbital, Configuration, and State Pictures of Electronic Structure One of the goals of quantum chemistry is to allow
4 Post-Hartree Fock Methods: MPn and Configuration Interaction
 4 Post-Hartree Fock Methods: MPn and Configuration Interaction In the limit of a complete basis, the Hartree-Fock (HF) energy in the complete basis set limit (ECBS HF ) yields an upper boundary to the
4 Post-Hartree Fock Methods: MPn and Configuration Interaction In the limit of a complete basis, the Hartree-Fock (HF) energy in the complete basis set limit (ECBS HF ) yields an upper boundary to the
Fully Automated Implementation of the Incremental Scheme: Application. to CCSD Energies for Hydrocarbons and Transition Metal Compounds
 Fully Automated Implementation of the Incremental Scheme: Application to CCSD Energies for Hydrocarbons and Transition Metal Compounds Joachim Friedrich 1, Michael Hanrath 1, Michael Dolg 1 1 Institute
Fully Automated Implementation of the Incremental Scheme: Application to CCSD Energies for Hydrocarbons and Transition Metal Compounds Joachim Friedrich 1, Michael Hanrath 1, Michael Dolg 1 1 Institute
Evaluation of a Characteristic Atomic Radius by an Ab Initio Method
 Evaluation of a Characteristic Atomic Radius by an Ab Initio Method ZHONG-ZHI YANG Department of Chemistry, Liaoning Normal University, Dalian, 116029, and Institute of Theoretical Chemistry, Jilin University,
Evaluation of a Characteristic Atomic Radius by an Ab Initio Method ZHONG-ZHI YANG Department of Chemistry, Liaoning Normal University, Dalian, 116029, and Institute of Theoretical Chemistry, Jilin University,
CHEM-UA 127: Advanced General Chemistry I
 CHEM-UA 7: Advanced General Chemistry I I. LINEAR COMBINATION OF ATOMIC ORBITALS Linear combination of atomic orbitals (LCAO) is a simple method of quantum chemistry that yields a qualitative picture of
CHEM-UA 7: Advanced General Chemistry I I. LINEAR COMBINATION OF ATOMIC ORBITALS Linear combination of atomic orbitals (LCAO) is a simple method of quantum chemistry that yields a qualitative picture of
Coupled-Cluster Perturbative Triples for Bond Breaking
 Coupled-Cluster Perturbative Triples for Bond Breaking Andrew G. Taube and Rodney J. Bartlett Quantum Theory Project University of Florida INT CC Meeting Seattle July 8, 2008 Why does chemistry need triples?
Coupled-Cluster Perturbative Triples for Bond Breaking Andrew G. Taube and Rodney J. Bartlett Quantum Theory Project University of Florida INT CC Meeting Seattle July 8, 2008 Why does chemistry need triples?
Kohn Sham density functional theory [1 3] is. Role of the Exchange Correlation Energy: Nature s Glue STEFAN KURTH, JOHN P. PERDEW.
![Kohn Sham density functional theory [1 3] is. Role of the Exchange Correlation Energy: Nature s Glue STEFAN KURTH, JOHN P. PERDEW.](/thumbs/80/82235561.jpg "Kohn Sham density functional theory [1 3] is. Role of the Exchange Correlation Energy: Nature s Glue STEFAN KURTH, JOHN P. PERDEW.") Role of the Exchange Correlation Energy: Nature s Glue STEFAN KURTH, JOHN P. PERDEW Department of Physics and Quantum Theory Group, Tulane University, New Orleans, Louisiana 70118 Received 11 March 1999;
Role of the Exchange Correlation Energy: Nature s Glue STEFAN KURTH, JOHN P. PERDEW Department of Physics and Quantum Theory Group, Tulane University, New Orleans, Louisiana 70118 Received 11 March 1999;
Potentials, periodicity
 Potentials, periodicity Lecture 2 1/23/18 1 Survey responses 2 Topic requests DFT (10), Molecular dynamics (7), Monte Carlo (5) Machine Learning (4), High-throughput, Databases (4) NEB, phonons, Non-equilibrium
Potentials, periodicity Lecture 2 1/23/18 1 Survey responses 2 Topic requests DFT (10), Molecular dynamics (7), Monte Carlo (5) Machine Learning (4), High-throughput, Databases (4) NEB, phonons, Non-equilibrium
Teoría del Funcional de la Densidad (Density Functional Theory)
") Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride. Dimer. Philip Straughn
 Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
equation of state and bulk modulus of AlP, AlAs and AlSb semiconductor compounds
 PRAMANA c Indian Academy of Sciences Vol. 64, No. 1 journal of January 2005 physics pp. 153 158 Total energy, equation of state and bulk modulus of AlP, AlAs and AlSb semiconductors A R JIVANI, H J TRIVEDI,
PRAMANA c Indian Academy of Sciences Vol. 64, No. 1 journal of January 2005 physics pp. 153 158 Total energy, equation of state and bulk modulus of AlP, AlAs and AlSb semiconductors A R JIVANI, H J TRIVEDI,
Quantum Condensed Matter Physics Lecture 4
 Quantum Condensed Matter Physics Lecture 4 David Ritchie QCMP Lent/Easter 2019 http://www.sp.phy.cam.ac.uk/drp2/home 4.1 Quantum Condensed Matter Physics 1. Classical and Semi-classical models for electrons
Quantum Condensed Matter Physics Lecture 4 David Ritchie QCMP Lent/Easter 2019 http://www.sp.phy.cam.ac.uk/drp2/home 4.1 Quantum Condensed Matter Physics 1. Classical and Semi-classical models for electrons
Static Dipole Moments and Electronic Structure Calculations of the Low-Lying Electronic States of the Molecule Zinc Selinum ZnSe
 Modern Applied Science; Vol. 11, No. 9; 2017 ISSN 1913-1844 E-ISSN 1913-1852 Published by Canadian Center of Science and Education Static Dipole Moments and Electronic Structure Calculations of the Low-Lying
Modern Applied Science; Vol. 11, No. 9; 2017 ISSN 1913-1844 E-ISSN 1913-1852 Published by Canadian Center of Science and Education Static Dipole Moments and Electronic Structure Calculations of the Low-Lying
Core exciton energies of bulk MgO, Al 2 O 3, and SiO 2 from explicitly correlated ab initio cluster model calculations
 PHYSICAL REVIEW B VOLUME 62, NUMBER 15 15 OCTOBER 2000-I Core exciton energies of bulk MgO, Al 2 O 3, and SiO 2 from explicitly correlated ab initio cluster model calculations Carmen Sousa, Coen de Graaf,
PHYSICAL REVIEW B VOLUME 62, NUMBER 15 15 OCTOBER 2000-I Core exciton energies of bulk MgO, Al 2 O 3, and SiO 2 from explicitly correlated ab initio cluster model calculations Carmen Sousa, Coen de Graaf,
1. Thomas-Fermi method
 1. Thomas-Fermi method We consider a system of N electrons in a stationary state, that would obey the stationary Schrödinger equation: h i m + 1 v(r i,r j ) Ψ(r 1,...,r N ) = E i Ψ(r 1,...,r N ). (1.1)
1. Thomas-Fermi method We consider a system of N electrons in a stationary state, that would obey the stationary Schrödinger equation: h i m + 1 v(r i,r j ) Ψ(r 1,...,r N ) = E i Ψ(r 1,...,r N ). (1.1)
Name: Date: Blk: Examine your periodic table to answer these questions and fill-in-the-blanks. Use drawings to support your answers where needed:
 Name: Date: Blk: NOTES: BONDING Examine your periodic table to answer these questions and fill-in-the-blanks. Use drawings to support your answers where needed: I. IONIC BONDING Ionic bond: formed by the
Name: Date: Blk: NOTES: BONDING Examine your periodic table to answer these questions and fill-in-the-blanks. Use drawings to support your answers where needed: I. IONIC BONDING Ionic bond: formed by the
Electron Correlation
 Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation
Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation