QSAR in Green Chemistry
|
|
|
- Estella West
- 5 years ago
- Views:
Transcription
1 QSAR in Green Chemistry Activity Relationship QSAR is the acronym for Quantitative Structure-Activity Relationship Chemistry is based on the premise that similar chemicals will behave similarly The behavior/activity of a chemical is derived from its structure QSAR research searches for relationships between chemical structure and activity Why QSAR? Limited toxicity data on majority of chemicals Priority setting for testing In QSAR, estimating behavior of untested chemicals has been used for >60 years
2 QSAR basics QSARs associated with endpoint ( i.e.lc50) QSAR attempt to group chemical behavior in terms of toxicity QSAR predicts biological activity (endpoints) directly from models of chemical structure Each QSAR predicts a specific endpoint Discrete Endpoints (e.g., Carcinogenicity, Mutagenicity) Chemical + Structure + Data SAR Software Toxicity Response Predictions
3 What a QSAR needs Set of chemicals Molecular Descriptors for chemicals Data Training and Test Model or Algorithm linking descriptors to endpoint Independent Validation (hopefully)

4 QSAR flow diagram

5

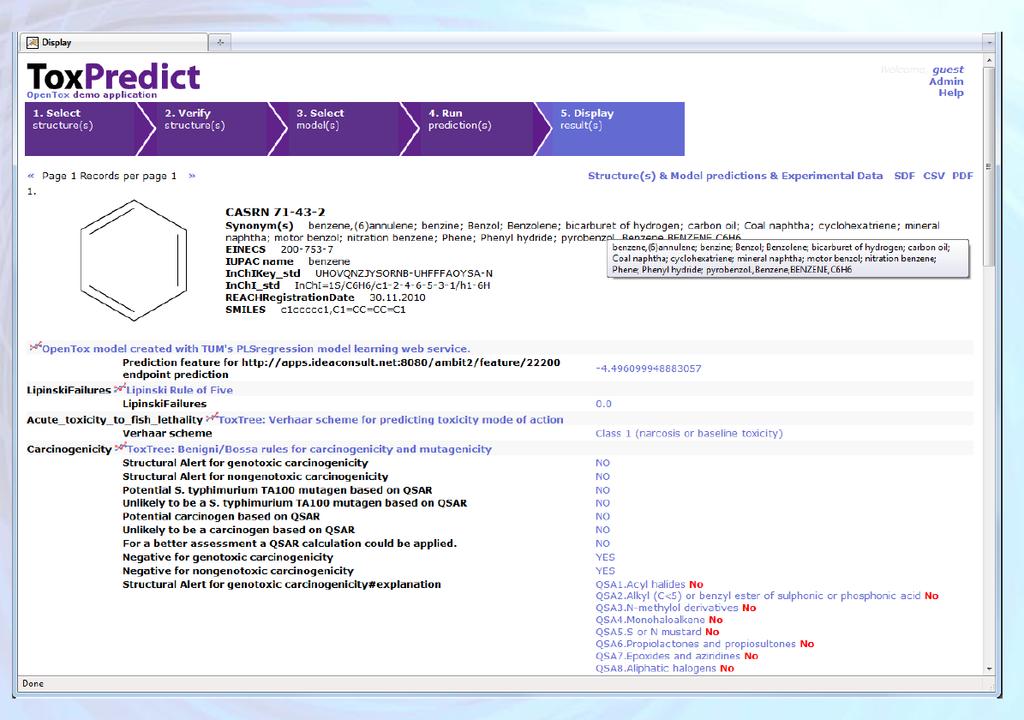
6 Open Tox Examples

7 Biomolecular Interactions Chemical Interactions DH f products metabolic transformation Intrinsic Chemical Reactivity Structure 2D 3D SurfArea chirality planarity volume distances pka functional groups fragments bonds electrostatic potential Toxicity MW Risk Assessment solvation energies H-bond energies electron affinities frontier orbital energies increasing model sophistication bond energies radical formation transition state energies rxn w/ model nuclephiles increasing prior knowledge energy of rxn intermediates
8 Molecular Descriptors Descriptors representing properties of complete molecules Descriptors calculated from 2D structure Descriptors requiring 3D representations
9 Descriptors representing properties of complete molecules SMILES-simplified molecular-input lineentry system -a machine-readable format -2D representation of molecules as linear strings of alpha-numeric characters. LogP, Hydrophobicity LogP the logarithm of the partition coefficient between n-octanol and water ClogP (Leo and Hansch) based on small set of values from a small set of simple molecules
10 Molecular Descriptors SMILES-simplified molecular-input line-entry system
11 Descriptors calculated from 2D structure Single-valued descriptors calculated from the 2D graph of the molecule Simple counts of features Lipinski Rule of Five (H bonds, MW, etc.) Number of ring systems Number of rotatable bonds Characterize structures according to size, degree of branching, connectivity and overall shape Wiener Index counts the number of bonds between pairs of atoms and sums the distances between all pairs Randić (et al.) branching index - Defines a degree of an atom as the number of adjacent non-hydrogen atoms Bond connectivity value is the reciprocal of the square root of the product of the degree of the two atoms in the bond. Branching index is the sum of the bond connectivities over all bonds in the molecule. Chi indexes introduces valence values to encode sigma, pi, and lone pair electrons Kappa Shape Indexes -characterize aspects of molecular shape Compare the molecule with the extreme shapes possible for that number of atoms Range from linear molecules to completely connected graph
12 2D fingerprints Nodes do not represent atoms but features such as functionally important groups or whole ring system.
13 DESCRIPTORS BASED ON 3D REPRESENTATIONS Require the generation of 3D conformations Can be computationally time consuming with large data sets Usually must take into account conformational flexibility 3D fragment screens encode spatial relationships between atoms, ring centroids, and planes Toxicophores Based on atoms or substructures thought to be relevant for toxicity Expert driven
14 DRAGON Dragon - an application for the calculation of molecular descriptors 1,600 molecular descriptors that are divided into 20 logical blocks. ACD Labs - ACD Labs values now incorporated into the CAS Registry File for millions of compounds I-Lab: Name generation NMR prediction Physical property prediction
15 Open Tox Descriptor Algorithms ToxDesc 11 descriptor calculation algorithms Existing methodss OpenBabel, JOELib2, The Chemistry Development Kit (CDK) and gspan, New methods FMiner, MakeMNA, and MakeQNA. AMBIT The single descriptor calculation web service developed by OpenTox partner IDEA offers descriptors calculated by several packages

16 Open Tox Descriptor Algorithms

17 Open Tox Descriptor Algorithms

18 Open Tox Descriptor Algorithms

19 Open Tox Descriptor Algorithms
20 Open Tox Data Sets Textual databases (e.g., IARC [35], NTP [36]); Machine readable files (e.g.,.sdf) DSSTox, ISSCAN, AMBIT,RepDose Public Databases PubChem, ACToR, CPDBAS, DBPCAN, EPAFHM, KIERBL, IRISTR, FDAMDD, ECETOC skin irritation, LLNA skin sensitization and the Bioconcentration factor (BCF) Gold Standard Database

21 Open Tox Dataset Selection

22 Open Tox Dataset Selection

23 Open Tox Dataset Selection
 Competitive binding assays for the estrogen nuclear")

24 Chemistry Space of the NCTR estrogen binding Dataset NCTR dataset: x Strong Weak inactive EPA dataset (58K) Competitive binding assays for the estrogen nuclear receptor proteins NCTR dataset cover a significant part of the chemistry space defined by the EPA dataset Most active compounds cluster together, whereas inactives scatter over the space.
25 Number of Chemicals Data sets NCTR Dataset (130 active and 100 inactive) RBA=100 for E Inactive < ~ ~ ~1 1~10 10~100 >100 Relative Binding Activity (RBA) Steroids DESs Phytoestrogens DDTs PCBs Alkyphenols phthalates pesticides others Over six orders of magnitude of RBA range A wide range of chemical classes
26 Algorithms Linear QSAR Models Supervised classification Unsupervised classification Clustering approaches Machine Learning methods Genetic Algorithms Neural Networks Self Organizing Maps (SOM) Toxicophore search
27 Open Tox Algorithms Feature Selection Algorithms -algorithms for the reduction of the dimensionality of a dataset, by selecting only a subset of a full set of descriptors included in the dataset including a partial least-squares filter, principle components analysis (PCA), chi-squared attribute evaluation, information-gain attribute evaluation, a scaling filter, and a missing value replacer; Un Supervised Clustering Algorithms Standard algorithms - unsupervised learning algorithms that group objects of similar kind into respective categories. TUM s StructuralClustering procedure Supervised Classification and Regression Algorithm several common (Q)SAR modelling algorithms (Linear Regression, Multiple Linear Regression, PLS, Gaussian Processes, Neural Networks) standard machine learning methods (SVMs, KNN, M5, J48, Naïve Bayes) in house algorithms from partners (lazar, ToxTree, BBRC, LastPM, LoMoGraph, MaxTox, isar, Three Conditional Density Estimators, MakeSCR) Applicability domain estimation algorithms is the chemical space adequately assessed
28 Linear QSAR models Simple linear model ypred is the model output, xi and wi the ith descriptor (out of a total of M) and corresponding coefficient (weight) respectively, and w0 an offset term Multiple linear Regression models Principal component regression models Partial Least squares regression Linear Discriminant analysis Naïve Bayes Aromatic amines mutagenicity model log TA100 = 0.92 Kow HOMO 1.18 LUMO log TA100 is the mutagenic potency (revertants/nmol) Kow hydrophobicity HOMO is the energy of the highest occupied molecular orbital LUMO is the energy of the lowest unoccupied molecular orbital.
29 Supervised Classification Approach Chemical Structures (represented by descriptors) Pattern recognition Pattern recognition methods: Classification and regression tree (CART) K-nearest neighbor (KNN) Artificial neural network (ANN) SIMCA Similar model results obtained using various pattern recognition methods Overall correct prediction rate is between 70-80% The nature of descriptors are more critical Active Inactive
30 CART Model Classification and Regression Tree (CART) > < Unique contribution to the overall prediction Descriptors are selected based on genetic algorithm clogp (hydrophobicity) Phenolic ring indicator Shadow-XY (length of structure) RPCS (relative positive charge surface area) PNSA (total charge weighted negative surface area CART uses a decision tree to display how chemical may be classified or predicted through a series of rules. These rules are expressed as if then..
31 Similarity Search Similarity Index Triazolam Cluster Identification Br Safety Data on the Chemical and Similar Substances Test Compound
32 Unsupervised clustering Compound Classification and Selection Agglomerative methods start at the bottom and merge similar clusters (bottom-up) Divisive hierarchical clustering starts with all compounds in a single cluster and partitions the data (top-down)
33 Machine Learning Methods Genetic algorithms Different parameters and model solutions to given problems are encoded in a chromosome and subjected to iterative random variation, thus generating a population. Solutions provided by these chromosomes are evaluated by fitness function that assign high scores to desired results. Chromosomes yielding best intermediate solutions are subjected to mutation and crossover operation that correspond to random genetic mutations and gene recombination events. The resulting modified chromosomes represent the next generation and the process is continued until the obtained results meet a satisfactory convergence criterion
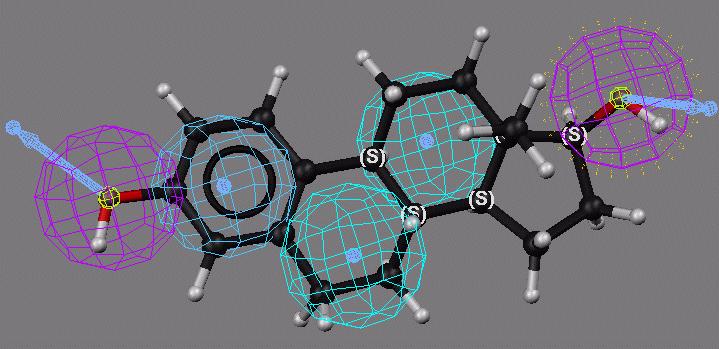



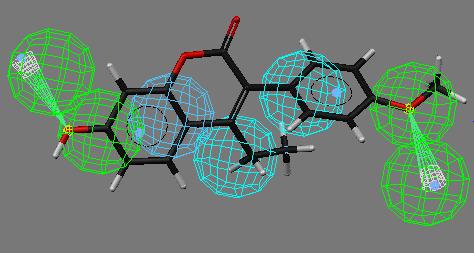
34 Toxicophore Searching Template Structure Query Hits Searching Match Database with multiple conformers
 Fragments NOT Exclusively Associated with Toxicity (~500,000, e.g.) Identity of Chemicals with Structural Alert Fragments Toxic Potency Number of Compounds / SA Presence and Identity of Modulators QSAR Properties Estimate Toxic Potential")
35 Estimating Toxic Potential Using toxicophore searching Reduce test molecule to all possible 2-10 atom fragments Compare molecular fragments to active and inactive fragments in the control toxicity database Fragments Exclusively Associated with Toxicity (~200, e.g.) Fragments NOT Exclusively Associated with Toxicity (~500,000, e.g.) Identity of Chemicals with Structural Alert Fragments Toxic Potency Number of Compounds / SA Presence and Identity of Modulators QSAR Properties Estimate Toxic Potential

36 Open Tox Model/Algorithm Selection

37 Open Tox Model/Algorithm Selection

38 Open Tox Model/Algorithm Selection
39 Assignment for next week Apply ToxPredict on your set of chemicals Focus on algorithms/models which use QSAR/SAR based predictions Compare QSAR predictions with Toxicophore based predictions Modify your compound in similar ways as last week Run QSAR predictions again Compare with Toxicophore based predictions
40 Validation False Positives Actual Negative Predicted Positive Predictive Value Predicted Positive Actual Positive Specificity Actual Negative Predicted Negative Sensitivity Actual Positive Predicted Positive
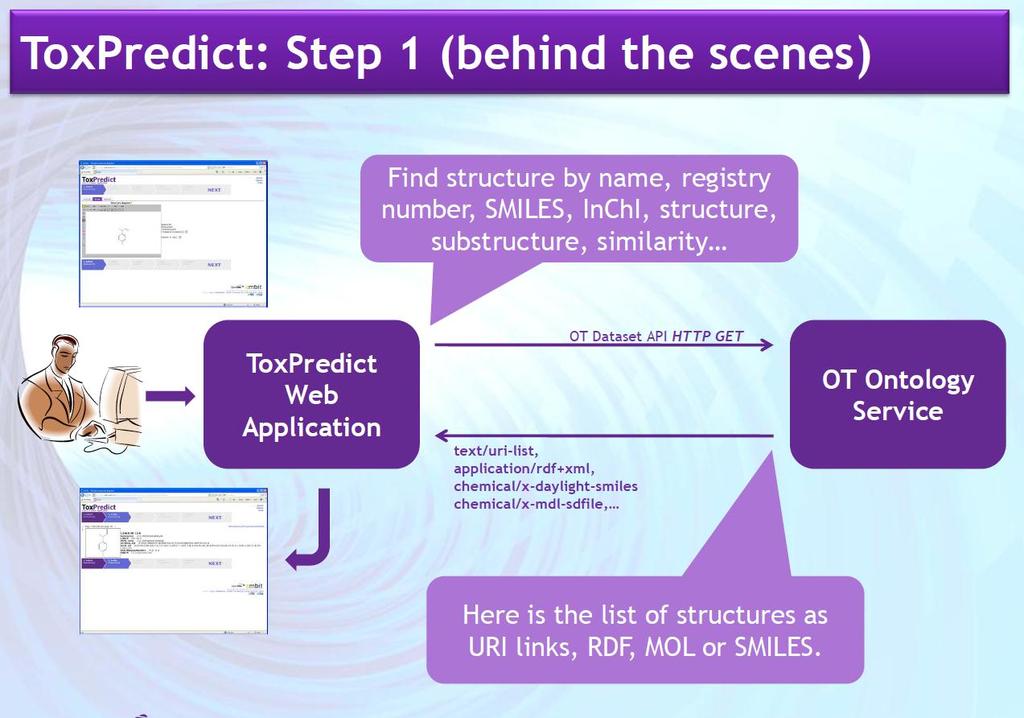
41

42
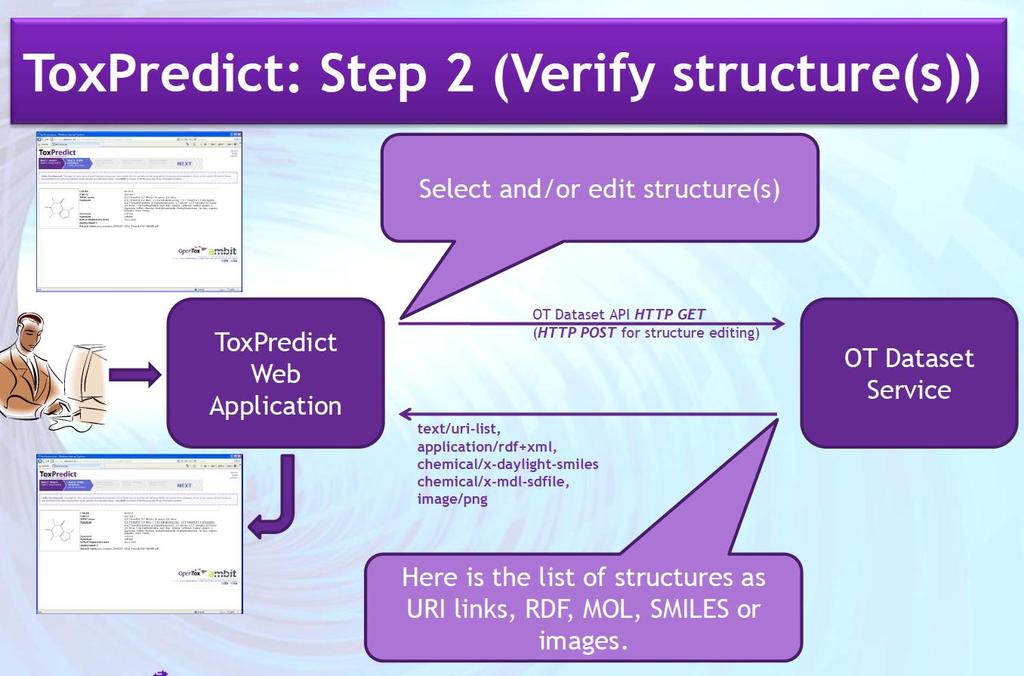
43
44

45
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Structure-Activity Modeling - QSAR. Uwe Koch
 Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
OECD QSAR Toolbox v.4.1. Tutorial on how to predict Skin sensitization potential taking into account alert performance
 OECD QSAR Toolbox v.4.1 Tutorial on how to predict Skin sensitization potential taking into account alert performance Outlook Background Objectives Specific Aims Read across and analogue approach The exercise
OECD QSAR Toolbox v.4.1 Tutorial on how to predict Skin sensitization potential taking into account alert performance Outlook Background Objectives Specific Aims Read across and analogue approach The exercise
Priority Setting of Endocrine Disruptors Using QSARs
 Priority Setting of Endocrine Disruptors Using QSARs Weida Tong Manager of Computational Science Group, Logicon ROW Sciences, FDA s National Center for Toxicological Research (NCTR), U.S.A. Thanks for
Priority Setting of Endocrine Disruptors Using QSARs Weida Tong Manager of Computational Science Group, Logicon ROW Sciences, FDA s National Center for Toxicological Research (NCTR), U.S.A. Thanks for
Quantitative Structure Activity Relationships: An overview
 Quantitative Structure Activity Relationships: An overview Prachi Pradeep Oak Ridge Institute for Science and Education Research Participant National Center for Computational Toxicology U.S. Environmental
Quantitative Structure Activity Relationships: An overview Prachi Pradeep Oak Ridge Institute for Science and Education Research Participant National Center for Computational Toxicology U.S. Environmental
OECD QSAR Toolbox v.4.0. Tutorial on how to predict Skin sensitization potential taking into account alert performance
 OECD QSAR Toolbox v.4.0 Tutorial on how to predict Skin sensitization potential taking into account alert performance Outlook Background Objectives Specific Aims Read across and analogue approach The exercise
OECD QSAR Toolbox v.4.0 Tutorial on how to predict Skin sensitization potential taking into account alert performance Outlook Background Objectives Specific Aims Read across and analogue approach The exercise
Plan. Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics.
 and its used in Chemoinformatics.") Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Data Mining in the Chemical Industry. Overview of presentation
 Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Quantitative Structure-Activity Relationship (QSAR) computational-drug-design.html
 computational-drug-design.html") Quantitative Structure-Activity Relationship (QSAR) http://www.biophys.mpg.de/en/theoretical-biophysics/ computational-drug-design.html 07.11.2017 Ahmad Reza Mehdipour 07.11.2017 Course Outline 1. 1.Ligand-
Quantitative Structure-Activity Relationship (QSAR) http://www.biophys.mpg.de/en/theoretical-biophysics/ computational-drug-design.html 07.11.2017 Ahmad Reza Mehdipour 07.11.2017 Course Outline 1. 1.Ligand-
Introduction to Chemoinformatics and Drug Discovery
 Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Machine Learning Concepts in Chemoinformatics
 Machine Learning Concepts in Chemoinformatics Martin Vogt B-IT Life Science Informatics Rheinische Friedrich-Wilhelms-Universität Bonn BigChem Winter School 2017 25. October Data Mining in Chemoinformatics
Machine Learning Concepts in Chemoinformatics Martin Vogt B-IT Life Science Informatics Rheinische Friedrich-Wilhelms-Universität Bonn BigChem Winter School 2017 25. October Data Mining in Chemoinformatics
OECD QSAR Toolbox v.4.1
 OECD QSAR Toolbox v.4.1 Step-by-step example on how to predict the skin sensitisation potential approach of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific
OECD QSAR Toolbox v.4.1 Step-by-step example on how to predict the skin sensitisation potential approach of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific
OECD QSAR Toolbox v.3.3
 OECD QSAR Toolbox v.3.3 Step-by-step example on how to predict the skin sensitisation potential of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific Aims Read
OECD QSAR Toolbox v.3.3 Step-by-step example on how to predict the skin sensitisation potential of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific Aims Read
OECD QSAR Toolbox v.4.1. Step-by-step example for predicting skin sensitization accounting for abiotic activation of chemicals
 OECD QSAR Toolbox v.4.1 Step-by-step example for predicting skin sensitization accounting for abiotic activation of chemicals Background Outlook Objectives The exercise Workflow 2 Background This is a
OECD QSAR Toolbox v.4.1 Step-by-step example for predicting skin sensitization accounting for abiotic activation of chemicals Background Outlook Objectives The exercise Workflow 2 Background This is a
OECD QSAR Toolbox v.3.4
 OECD QSAR Toolbox v.3.4 Step-by-step example on how to predict the skin sensitisation potential approach of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific
OECD QSAR Toolbox v.3.4 Step-by-step example on how to predict the skin sensitisation potential approach of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific
QMRF# Title. number and title in JRC QSAR Model Data base 2.0 (new) number and title in JRC QSAR Model Data base 1.0
 number and title in JRC QSAR Model Data base 1.0") Q15-410-0003 ACD/Percepta model for genotoxicity (Ames test) Q31-47-42-424 ACD/Percepta model for genotoxicity (Ames test) Q15-42-0005 ACD/Percepta model for mouse acute oral toxicity Q32-48-43-426 ACD/Percepta
Q15-410-0003 ACD/Percepta model for genotoxicity (Ames test) Q31-47-42-424 ACD/Percepta model for genotoxicity (Ames test) Q15-42-0005 ACD/Percepta model for mouse acute oral toxicity Q32-48-43-426 ACD/Percepta
An Integrated Approach to in-silico
 An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
OECD QSAR Toolbox v.3.3. Predicting skin sensitisation potential of a chemical using skin sensitization data extracted from ECHA CHEM database
 OECD QSAR Toolbox v.3.3 Predicting skin sensitisation potential of a chemical using skin sensitization data extracted from ECHA CHEM database Outlook Background The exercise Workflow Save prediction 23.02.2015
OECD QSAR Toolbox v.3.3 Predicting skin sensitisation potential of a chemical using skin sensitization data extracted from ECHA CHEM database Outlook Background The exercise Workflow Save prediction 23.02.2015
Computational Methods and Drug-Likeness. Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004
 Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
Screening and prioritisation of substances of concern: A regulators perspective within the JANUS project
 Für Mensch & Umwelt LIFE COMBASE workshop on Computational Tools for the Assessment and Substitution of Biocidal Active Substances of Ecotoxicological Concern Screening and prioritisation of substances
Für Mensch & Umwelt LIFE COMBASE workshop on Computational Tools for the Assessment and Substitution of Biocidal Active Substances of Ecotoxicological Concern Screening and prioritisation of substances
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Development of a Structure Generator to Explore Target Areas on Chemical Space
 Development of a Structure Generator to Explore Target Areas on Chemical Space Kimito Funatsu Department of Chemical System Engineering, This materials will be published on Molecular Informatics Drug Development
Development of a Structure Generator to Explore Target Areas on Chemical Space Kimito Funatsu Department of Chemical System Engineering, This materials will be published on Molecular Informatics Drug Development
OECD QSAR Toolbox v.3.4. Example for predicting Repeated dose toxicity of 2,3-dimethylaniline
 OECD QSAR Toolbox v.3.4 Example for predicting Repeated dose toxicity of 2,3-dimethylaniline Outlook Background Objectives The exercise Workflow Save prediction 2 Background This is a step-by-step presentation
OECD QSAR Toolbox v.3.4 Example for predicting Repeated dose toxicity of 2,3-dimethylaniline Outlook Background Objectives The exercise Workflow Save prediction 2 Background This is a step-by-step presentation
OECD QSAR Toolbox v.3.4
 OECD QSAR Toolbox v.3.4 Predicting developmental and reproductive toxicity of Diuron (CAS 330-54-1) based on DART categorization tool and DART SAR model Outlook Background Objectives The exercise Workflow
OECD QSAR Toolbox v.3.4 Predicting developmental and reproductive toxicity of Diuron (CAS 330-54-1) based on DART categorization tool and DART SAR model Outlook Background Objectives The exercise Workflow
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
KATE2017 on NET beta version https://kate2.nies.go.jp/nies/ Operating manual
 KATE2017 on NET beta version http://kate.nies.go.jp https://kate2.nies.go.jp/nies/ Operating manual 2018.03.29 KATE2017 on NET was developed to predict the following ecotoxicity values: 50% effective concentration
KATE2017 on NET beta version http://kate.nies.go.jp https://kate2.nies.go.jp/nies/ Operating manual 2018.03.29 KATE2017 on NET was developed to predict the following ecotoxicity values: 50% effective concentration
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
OECD QSAR Toolbox v.3.2. Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding
 OECD QSAR Toolbox v.3.2 Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding Outlook Background Objectives Specific Aims The exercise Workflow
OECD QSAR Toolbox v.3.2 Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding Outlook Background Objectives Specific Aims The exercise Workflow
OECD QSAR Toolbox v.3.4. Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding
 OECD QSAR Toolbox v.3.4 Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding Outlook Background Objectives Specific Aims The exercise Workflow
OECD QSAR Toolbox v.3.4 Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding Outlook Background Objectives Specific Aims The exercise Workflow
OECD QSAR Toolbox v.3.3. Step-by-step example of how to build a userdefined
 OECD QSAR Toolbox v.3.3 Step-by-step example of how to build a userdefined QSAR Background Objectives The exercise Workflow of the exercise Outlook 2 Background This is a step-by-step presentation designed
OECD QSAR Toolbox v.3.3 Step-by-step example of how to build a userdefined QSAR Background Objectives The exercise Workflow of the exercise Outlook 2 Background This is a step-by-step presentation designed
OECD QSAR Toolbox v.4.1. Step-by-step example for building QSAR model
 OECD QSAR Toolbox v.4.1 Step-by-step example for building QSAR model Background Objectives The exercise Workflow of the exercise Outlook 2 Background This is a step-by-step presentation designed to take
OECD QSAR Toolbox v.4.1 Step-by-step example for building QSAR model Background Objectives The exercise Workflow of the exercise Outlook 2 Background This is a step-by-step presentation designed to take
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
OECD QSAR Toolbox v.3.3. Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding
 OECD QSAR Toolbox v.3.3 Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding Outlook Background Objectives Specific Aims The exercise Workflow
OECD QSAR Toolbox v.3.3 Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding Outlook Background Objectives Specific Aims The exercise Workflow
OECD QSAR Toolbox v.3.0
 OECD QSAR Toolbox v.3.0 Step-by-step example of how to categorize an inventory by mechanistic behaviour of the chemicals which it consists Background Objectives Specific Aims Trend analysis The exercise
OECD QSAR Toolbox v.3.0 Step-by-step example of how to categorize an inventory by mechanistic behaviour of the chemicals which it consists Background Objectives Specific Aims Trend analysis The exercise
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
OECD QSAR Toolbox v.3.3. Step-by-step example of how to categorize an inventory by mechanistic behaviour of the chemicals which it consists
 OECD QSAR Toolbox v.3.3 Step-by-step example of how to categorize an inventory by mechanistic behaviour of the chemicals which it consists Background Objectives Specific Aims Trend analysis The exercise
OECD QSAR Toolbox v.3.3 Step-by-step example of how to categorize an inventory by mechanistic behaviour of the chemicals which it consists Background Objectives Specific Aims Trend analysis The exercise
QSAR Modeling of Human Liver Microsomal Stability Alexey Zakharov
 QSAR Modeling of Human Liver Microsomal Stability Alexey Zakharov CADD Group Chemical Biology Laboratory Frederick National Laboratory for Cancer Research National Cancer Institute, National Institutes
QSAR Modeling of Human Liver Microsomal Stability Alexey Zakharov CADD Group Chemical Biology Laboratory Frederick National Laboratory for Cancer Research National Cancer Institute, National Institutes
Chapter 8: Introduction to QSAR
 : Introduction to 8) Chapter 8: 181 8.1 Introduction to 181 8.2 Objectives of 181 8.3 Historical development of 182 8.4 Molecular descriptors used in 183 8.5 Methods of 185 8.5.1 2D methods 186 8.6 Introduction
: Introduction to 8) Chapter 8: 181 8.1 Introduction to 181 8.2 Objectives of 181 8.3 Historical development of 182 8.4 Molecular descriptors used in 183 8.5 Methods of 185 8.5.1 2D methods 186 8.6 Introduction
COMPUTER AIDED DRUG DESIGN (CADD) AND DEVELOPMENT METHODS
 AND DEVELOPMENT METHODS") COMPUTER AIDED DRUG DESIGN (CADD) AND DEVELOPMENT METHODS DRUG DEVELOPMENT Drug development is a challenging path Today, the causes of many diseases (rheumatoid arthritis, cancer, mental diseases, etc.)
COMPUTER AIDED DRUG DESIGN (CADD) AND DEVELOPMENT METHODS DRUG DEVELOPMENT Drug development is a challenging path Today, the causes of many diseases (rheumatoid arthritis, cancer, mental diseases, etc.)
Using Self-Organizing maps to accelerate similarity search
 YOU LOGO Using Self-Organizing maps to accelerate similarity search Fanny Bonachera, Gilles Marcou, Natalia Kireeva, Alexandre Varnek, Dragos Horvath Laboratoire d Infochimie, UM 7177. 1, rue Blaise Pascal,
YOU LOGO Using Self-Organizing maps to accelerate similarity search Fanny Bonachera, Gilles Marcou, Natalia Kireeva, Alexandre Varnek, Dragos Horvath Laboratoire d Infochimie, UM 7177. 1, rue Blaise Pascal,
OECD QSAR Toolbox v.4.1. Tutorial illustrating new options for grouping with metabolism
 OECD QSAR Toolbox v.4.1 Tutorial illustrating new options for grouping with metabolism Outlook Background Objectives Specific Aims The exercise Workflow 2 Background Grouping with metabolism is a procedure
OECD QSAR Toolbox v.4.1 Tutorial illustrating new options for grouping with metabolism Outlook Background Objectives Specific Aims The exercise Workflow 2 Background Grouping with metabolism is a procedure
OECD QSAR Toolbox v.3.3. Predicting acute aquatic toxicity to fish of Dodecanenitrile (CAS ) taking into account tautomerism
 taking into account tautomerism") OECD QSAR Toolbox v.3.3 Predicting acute aquatic toxicity to fish of Dodecanenitrile (CAS 2437-25-4) taking into account tautomerism Outlook Background Objectives The exercise Workflow Save prediction
OECD QSAR Toolbox v.3.3 Predicting acute aquatic toxicity to fish of Dodecanenitrile (CAS 2437-25-4) taking into account tautomerism Outlook Background Objectives The exercise Workflow Save prediction
Read-Across or QSARs?
 Replacing Experimentation Read-Across or QSARs? Which one to apply and when? Presented by: Dr. Faizan SAHIGARA Chemical Watch Expo 2017 26th April, 2017 Berlin Germany KREATiS, 23 rue du creuzat, 38080
Replacing Experimentation Read-Across or QSARs? Which one to apply and when? Presented by: Dr. Faizan SAHIGARA Chemical Watch Expo 2017 26th April, 2017 Berlin Germany KREATiS, 23 rue du creuzat, 38080
molecules ISSN
 Molecules 2004, 9, 1004-1009 molecules ISSN 1420-3049 http://www.mdpi.org Performance of Kier-Hall E-state Descriptors in Quantitative Structure Activity Relationship (QSAR) Studies of Multifunctional
Molecules 2004, 9, 1004-1009 molecules ISSN 1420-3049 http://www.mdpi.org Performance of Kier-Hall E-state Descriptors in Quantitative Structure Activity Relationship (QSAR) Studies of Multifunctional
HYPERGRAPH BASED SEMI-SUPERVISED LEARNING ALGORITHMS APPLIED TO SPEECH RECOGNITION PROBLEM: A NOVEL APPROACH
 HYPERGRAPH BASED SEMI-SUPERVISED LEARNING ALGORITHMS APPLIED TO SPEECH RECOGNITION PROBLEM: A NOVEL APPROACH Hoang Trang 1, Tran Hoang Loc 1 1 Ho Chi Minh City University of Technology-VNU HCM, Ho Chi
HYPERGRAPH BASED SEMI-SUPERVISED LEARNING ALGORITHMS APPLIED TO SPEECH RECOGNITION PROBLEM: A NOVEL APPROACH Hoang Trang 1, Tran Hoang Loc 1 1 Ho Chi Minh City University of Technology-VNU HCM, Ho Chi
Condensed Graph of Reaction: considering a chemical reaction as one single pseudo molecule
 Condensed Graph of Reaction: considering a chemical reaction as one single pseudo molecule Frank Hoonakker 1,3, Nicolas Lachiche 2, Alexandre Varnek 3, and Alain Wagner 3,4 1 Chemoinformatics laboratory,
Condensed Graph of Reaction: considering a chemical reaction as one single pseudo molecule Frank Hoonakker 1,3, Nicolas Lachiche 2, Alexandre Varnek 3, and Alain Wagner 3,4 1 Chemoinformatics laboratory,
Holdout and Cross-Validation Methods Overfitting Avoidance
 Holdout and Cross-Validation Methods Overfitting Avoidance Decision Trees Reduce error pruning Cost-complexity pruning Neural Networks Early stopping Adjusting Regularizers via Cross-Validation Nearest
Holdout and Cross-Validation Methods Overfitting Avoidance Decision Trees Reduce error pruning Cost-complexity pruning Neural Networks Early stopping Adjusting Regularizers via Cross-Validation Nearest
Course in Data Science
 Course in Data Science About the Course: In this course you will get an introduction to the main tools and ideas which are required for Data Scientist/Business Analyst/Data Analyst. The course gives an
Course in Data Science About the Course: In this course you will get an introduction to the main tools and ideas which are required for Data Scientist/Business Analyst/Data Analyst. The course gives an
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
PATTERN CLASSIFICATION
 PATTERN CLASSIFICATION Second Edition Richard O. Duda Peter E. Hart David G. Stork A Wiley-lnterscience Publication JOHN WILEY & SONS, INC. New York Chichester Weinheim Brisbane Singapore Toronto CONTENTS
PATTERN CLASSIFICATION Second Edition Richard O. Duda Peter E. Hart David G. Stork A Wiley-lnterscience Publication JOHN WILEY & SONS, INC. New York Chichester Weinheim Brisbane Singapore Toronto CONTENTS
Notes of Dr. Anil Mishra at 1
 Introduction Quantitative Structure-Activity Relationships QSPR Quantitative Structure-Property Relationships What is? is a mathematical relationship between a biological activity of a molecular system
Introduction Quantitative Structure-Activity Relationships QSPR Quantitative Structure-Property Relationships What is? is a mathematical relationship between a biological activity of a molecular system
bcl::cheminfo Suite Enables Machine Learning-Based Drug Discovery Using GPUs Edward W. Lowe, Jr. Nils Woetzel May 17, 2012
 bcl::cheminfo Suite Enables Machine Learning-Based Drug Discovery Using GPUs Edward W. Lowe, Jr. Nils Woetzel May 17, 2012 Outline Machine Learning Cheminformatics Framework QSPR logp QSAR mglur 5 CYP
bcl::cheminfo Suite Enables Machine Learning-Based Drug Discovery Using GPUs Edward W. Lowe, Jr. Nils Woetzel May 17, 2012 Outline Machine Learning Cheminformatics Framework QSPR logp QSAR mglur 5 CYP
QSAR APPLICATION TOOLBOX ADVANCED TRAINING WORKSHOP. BARCELONA, SPAIN 3-4, June 2015 AGENDA
 QSAR APPLICATION TOOLBOX ADVANCED TRAINING WORKSHOP BARCELONA, SPAIN 3-4, June 2015 AGENDA Wednesday, 3 June 2015 (09:00 17:30) 09:00-09:15 Registration Welcome and Introductions/Announcements 09:15-10:00
QSAR APPLICATION TOOLBOX ADVANCED TRAINING WORKSHOP BARCELONA, SPAIN 3-4, June 2015 AGENDA Wednesday, 3 June 2015 (09:00 17:30) 09:00-09:15 Registration Welcome and Introductions/Announcements 09:15-10:00
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics
 Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
A Comparison of Probabilistic, Neural, and Fuzzy Modeling in Ecotoxicity
 A Comparison of Probabilistic, Neural, and Fuzzy Modeling in Ecotoxicity Giuseppina Gini, Marco Giumelli DEI, Politecnico di Milano, piazza L. da Vinci 32, Milan, Italy Emilio Benfenati, Nadège Piclin
A Comparison of Probabilistic, Neural, and Fuzzy Modeling in Ecotoxicity Giuseppina Gini, Marco Giumelli DEI, Politecnico di Milano, piazza L. da Vinci 32, Milan, Italy Emilio Benfenati, Nadège Piclin
Exploring the black box: structural and functional interpretation of QSAR models.
 EMBL-EBI Industry workshop: In Silico ADMET prediction 4-5 December 2014, Hinxton, UK Exploring the black box: structural and functional interpretation of QSAR models. (Automatic exploration of datasets
EMBL-EBI Industry workshop: In Silico ADMET prediction 4-5 December 2014, Hinxton, UK Exploring the black box: structural and functional interpretation of QSAR models. (Automatic exploration of datasets
Microarray Data Analysis: Discovery
 Microarray Data Analysis: Discovery Lecture 5 Classification Classification vs. Clustering Classification: Goal: Placing objects (e.g. genes) into meaningful classes Supervised Clustering: Goal: Discover
Microarray Data Analysis: Discovery Lecture 5 Classification Classification vs. Clustering Classification: Goal: Placing objects (e.g. genes) into meaningful classes Supervised Clustering: Goal: Discover
Regulatory use of (Q)SARs under REACH
SARs under REACH") Regulatory use of (Q)SARs under REACH Webinar on Information requirements 10 December 2009 http://echa.europa.eu 1 Using (Q)SAR models Application under REACH to fulfill information requirements Use of
Regulatory use of (Q)SARs under REACH Webinar on Information requirements 10 December 2009 http://echa.europa.eu 1 Using (Q)SAR models Application under REACH to fulfill information requirements Use of
Navigation in Chemical Space Towards Biological Activity. Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland
 Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Modern Information Retrieval
 Modern Information Retrieval Chapter 8 Text Classification Introduction A Characterization of Text Classification Unsupervised Algorithms Supervised Algorithms Feature Selection or Dimensionality Reduction
Modern Information Retrieval Chapter 8 Text Classification Introduction A Characterization of Text Classification Unsupervised Algorithms Supervised Algorithms Feature Selection or Dimensionality Reduction
Principles of Pattern Recognition. C. A. Murthy Machine Intelligence Unit Indian Statistical Institute Kolkata
 Principles of Pattern Recognition C. A. Murthy Machine Intelligence Unit Indian Statistical Institute Kolkata e-mail: murthy@isical.ac.in Pattern Recognition Measurement Space > Feature Space >Decision
Principles of Pattern Recognition C. A. Murthy Machine Intelligence Unit Indian Statistical Institute Kolkata e-mail: murthy@isical.ac.in Pattern Recognition Measurement Space > Feature Space >Decision
Convolutional Embedding of Attributed Molecular Graphs for Physical Property Prediction
 Convolutional Embedding of Attributed Molecular Graphs for Physical Property Prediction The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters.
Convolutional Embedding of Attributed Molecular Graphs for Physical Property Prediction The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters.
QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression
 APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor
APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor
Plan. Day 2: Exercise on MHC molecules.
 Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Text Mining. Dr. Yanjun Li. Associate Professor. Department of Computer and Information Sciences Fordham University
 Text Mining Dr. Yanjun Li Associate Professor Department of Computer and Information Sciences Fordham University Outline Introduction: Data Mining Part One: Text Mining Part Two: Preprocessing Text Data
Text Mining Dr. Yanjun Li Associate Professor Department of Computer and Information Sciences Fordham University Outline Introduction: Data Mining Part One: Text Mining Part Two: Preprocessing Text Data
DISCOVERING new drugs is an expensive and challenging
 1036 IEEE TRANSACTIONS ON KNOWLEDGE AND DATA ENGINEERING, VOL. 17, NO. 8, AUGUST 2005 Frequent Substructure-Based Approaches for Classifying Chemical Compounds Mukund Deshpande, Michihiro Kuramochi, Nikil
1036 IEEE TRANSACTIONS ON KNOWLEDGE AND DATA ENGINEERING, VOL. 17, NO. 8, AUGUST 2005 Frequent Substructure-Based Approaches for Classifying Chemical Compounds Mukund Deshpande, Michihiro Kuramochi, Nikil
A Review on Computational Methods in Developing Quantitative Structure-Activity Relationship (QSAR)
") Navdeep Singh Sethi: A Review on Computational Methods in Developing Quantitative Structure-Activity 815 International Journal of Drug Design and Discovery Volume 3 Issue 3 July September 2012. 815-836
Navdeep Singh Sethi: A Review on Computational Methods in Developing Quantitative Structure-Activity 815 International Journal of Drug Design and Discovery Volume 3 Issue 3 July September 2012. 815-836
The In Silico Model for Mutagenicity
 Inn o vat i v e te c h n o l o g i e s, c o n c e p t s an d ap p r o a c h e s The In Silico Model for Mutagenicity Giuseppina Gini 1, Thomas Ferrari 1 and Alessandra Roncaglioni 2 1 DEI, Politecnico
Inn o vat i v e te c h n o l o g i e s, c o n c e p t s an d ap p r o a c h e s The In Silico Model for Mutagenicity Giuseppina Gini 1, Thomas Ferrari 1 and Alessandra Roncaglioni 2 1 DEI, Politecnico
ECO24 Prediction of Non-Extractable Residues Using Structural Information ( Structural Alerts )
") ECO24 Prediction of Non-Extractable Residues Using Structural Information ( Structural Alerts ) Ralph Kühne 1 Anja Miltner 2, Matthias Kästner 2, Norbert Ost 1, Andreas Schäffer 3, Gerrit Schüürmann 1,4
ECO24 Prediction of Non-Extractable Residues Using Structural Information ( Structural Alerts ) Ralph Kühne 1 Anja Miltner 2, Matthias Kästner 2, Norbert Ost 1, Andreas Schäffer 3, Gerrit Schüürmann 1,4
#33 - Genomics 11/09/07
 BCB 444/544 Required Reading (before lecture) Lecture 33 Mon Nov 5 - Lecture 31 Phylogenetics Parsimony and ML Chp 11 - pp 142 169 Genomics Wed Nov 7 - Lecture 32 Machine Learning Fri Nov 9 - Lecture 33
BCB 444/544 Required Reading (before lecture) Lecture 33 Mon Nov 5 - Lecture 31 Phylogenetics Parsimony and ML Chp 11 - pp 142 169 Genomics Wed Nov 7 - Lecture 32 Machine Learning Fri Nov 9 - Lecture 33
In Silico Investigation of Off-Target Effects
 PHARMA & LIFE SCIENCES WHITEPAPER In Silico Investigation of Off-Target Effects STREAMLINING IN SILICO PROFILING In silico techniques require exhaustive data and sophisticated, well-structured informatics
PHARMA & LIFE SCIENCES WHITEPAPER In Silico Investigation of Off-Target Effects STREAMLINING IN SILICO PROFILING In silico techniques require exhaustive data and sophisticated, well-structured informatics
AMBIT. Modules. Database. Chemical compounds. Table Error! No text of specified style in document..1
 AMBIT AMBIT is open source software for chemoinformatics data management developed with funding industry via CEFIC LRI funded project. AMBIT2 software consists of a database and functional modules allowing
AMBIT AMBIT is open source software for chemoinformatics data management developed with funding industry via CEFIC LRI funded project. AMBIT2 software consists of a database and functional modules allowing
OECD QSAR Toolbox v.3.3
 OECD QSAR Toolbox v.3.3 Step-by-step example of how to predict aquatic toxicity to Tetrahymena pyriformis by trend analysis using category pruning capabilities Outlook Background Objectives Specific Aims
OECD QSAR Toolbox v.3.3 Step-by-step example of how to predict aquatic toxicity to Tetrahymena pyriformis by trend analysis using category pruning capabilities Outlook Background Objectives Specific Aims
OECD QSAR Toolbox v.4.1. Tutorial illustrating new options of the structure similarity
 OECD QSAR Toolbox v.4.1 Tutorial illustrating new options of the structure similarity Outlook Background Aims PubChem features The exercise Workflow 2 Background This presentation is designed to familiarize
OECD QSAR Toolbox v.4.1 Tutorial illustrating new options of the structure similarity Outlook Background Aims PubChem features The exercise Workflow 2 Background This presentation is designed to familiarize
Representation of molecular structures. Coutersy of Prof. João Aires-de-Sousa, University of Lisbon, Portugal
 Representation of molecular structures Coutersy of Prof. João Aires-de-Sousa, University of Lisbon, Portugal A hierarchy of structure representations Name (S)-Tryptophan 2D Structure 3D Structure Molecular
Representation of molecular structures Coutersy of Prof. João Aires-de-Sousa, University of Lisbon, Portugal A hierarchy of structure representations Name (S)-Tryptophan 2D Structure 3D Structure Molecular
Translating Methods from Pharma to Flavours & Fragrances
 Translating Methods from Pharma to Flavours & Fragrances CINF 27: ACS National Meeting, New Orleans, LA - 18 th March 2018 Peter Hunt, Edmund Champness, Nicholas Foster, Tamsin Mansley & Matthew Segall
Translating Methods from Pharma to Flavours & Fragrances CINF 27: ACS National Meeting, New Orleans, LA - 18 th March 2018 Peter Hunt, Edmund Champness, Nicholas Foster, Tamsin Mansley & Matthew Segall
Statistical concepts in QSAR.
 Statistical concepts in QSAR. Computational chemistry represents molecular structures as a numerical models and simulates their behavior with the equations of quantum and classical physics. Available programs
Statistical concepts in QSAR. Computational chemistry represents molecular structures as a numerical models and simulates their behavior with the equations of quantum and classical physics. Available programs
Quantitative structure activity relationship and drug design: A Review
 International Journal of Research in Biosciences Vol. 5 Issue 4, pp. (1-5), October 2016 Available online at http://www.ijrbs.in ISSN 2319-2844 Research Paper Quantitative structure activity relationship
International Journal of Research in Biosciences Vol. 5 Issue 4, pp. (1-5), October 2016 Available online at http://www.ijrbs.in ISSN 2319-2844 Research Paper Quantitative structure activity relationship
Machine learning comes from Bayesian decision theory in statistics. There we want to minimize the expected value of the loss function.
 Bayesian learning: Machine learning comes from Bayesian decision theory in statistics. There we want to minimize the expected value of the loss function. Let y be the true label and y be the predicted
Bayesian learning: Machine learning comes from Bayesian decision theory in statistics. There we want to minimize the expected value of the loss function. Let y be the true label and y be the predicted
CHAPTER 6 QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIP (QSAR) ANALYSIS
 ANALYSIS") 159 CHAPTER 6 QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIP (QSAR) ANALYSIS 6.1 INTRODUCTION The purpose of this study is to gain on insight into structural features related the anticancer, antioxidant
159 CHAPTER 6 QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIP (QSAR) ANALYSIS 6.1 INTRODUCTION The purpose of this study is to gain on insight into structural features related the anticancer, antioxidant
Computational Approaches towards Life Detection by Mass Spectrometry
 Markus Meringer Computational Approaches towards Life Detection by Mass Spectrometry International Workshop on Life Detection Technology: For Mars, Encheladus and Beyond Earth-Life Science Institute, Tokyo
Markus Meringer Computational Approaches towards Life Detection by Mass Spectrometry International Workshop on Life Detection Technology: For Mars, Encheladus and Beyond Earth-Life Science Institute, Tokyo
Final Exam, Machine Learning, Spring 2009
 Name: Andrew ID: Final Exam, 10701 Machine Learning, Spring 2009 - The exam is open-book, open-notes, no electronics other than calculators. - The maximum possible score on this exam is 100. You have 3
Name: Andrew ID: Final Exam, 10701 Machine Learning, Spring 2009 - The exam is open-book, open-notes, no electronics other than calculators. - The maximum possible score on this exam is 100. You have 3
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Chemoinformatics and information management. Peter Willett, University of Sheffield, UK
 Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
1.QSAR identifier 1.1.QSAR identifier (title): QSAR for Relative Binding Affinity to Estrogen Receptor 1.2.Other related models:
: QSAR for Relative Binding Affinity to Estrogen Receptor 1.2.Other related models:") QMRF identifier (ECB Inventory): QMRF Title: QSAR for Relative Binding Affinity to Estrogen Receptor Printing Date:Feb 16, 2010 1.QSAR identifier 1.1.QSAR identifier (title): QSAR for Relative Binding
QMRF identifier (ECB Inventory): QMRF Title: QSAR for Relative Binding Affinity to Estrogen Receptor Printing Date:Feb 16, 2010 1.QSAR identifier 1.1.QSAR identifier (title): QSAR for Relative Binding
ChemAxon. Content. By György Pirok. D Standardization D Virtual Reactions. D Fragmentation. ChemAxon European UGM Visegrad 2008
 Transformers f off ChemAxon By György Pirok Content Standardization Virtual Reactions Metabolism M b li P Prediction di i Fragmentation 2 1 Standardization http://www.chemaxon.com/jchem/doc/user/standardizer.html
Transformers f off ChemAxon By György Pirok Content Standardization Virtual Reactions Metabolism M b li P Prediction di i Fragmentation 2 1 Standardization http://www.chemaxon.com/jchem/doc/user/standardizer.html
ARTIFICIAL NEURAL NETWORKS گروه مطالعاتي 17 بهار 92
 ARTIFICIAL NEURAL NETWORKS گروه مطالعاتي 17 بهار 92 BIOLOGICAL INSPIRATIONS Some numbers The human brain contains about 10 billion nerve cells (neurons) Each neuron is connected to the others through 10000
ARTIFICIAL NEURAL NETWORKS گروه مطالعاتي 17 بهار 92 BIOLOGICAL INSPIRATIONS Some numbers The human brain contains about 10 billion nerve cells (neurons) Each neuron is connected to the others through 10000
Coefficient Symbol Equation Limits
 1 Coefficient Symbol Equation Limits Squared Correlation Coefficient R 2 or r 2 0 r 2 N 1 2 ( Yexp, i Ycalc, i ) 2 ( Yexp, i Y ) i= 1 2 Cross-Validated R 2 q 2 r 2 or Q 2 or q 2 N 2 ( Yexp, i Ypred, i
1 Coefficient Symbol Equation Limits Squared Correlation Coefficient R 2 or r 2 0 r 2 N 1 2 ( Yexp, i Ycalc, i ) 2 ( Yexp, i Y ) i= 1 2 Cross-Validated R 2 q 2 r 2 or Q 2 or q 2 N 2 ( Yexp, i Ypred, i
QSAR/QSPR modeling. Quantitative Structure-Activity Relationships Quantitative Structure-Property-Relationships
 Quantitative Structure-Activity Relationships Quantitative Structure-Property-Relationships QSAR/QSPR modeling Alexandre Varnek Faculté de Chimie, ULP, Strasbourg, FRANCE QSAR/QSPR models Development Validation
Quantitative Structure-Activity Relationships Quantitative Structure-Property-Relationships QSAR/QSPR modeling Alexandre Varnek Faculté de Chimie, ULP, Strasbourg, FRANCE QSAR/QSPR models Development Validation
Chemical Space: Modeling Exploration & Understanding
 verview Chemical Space: Modeling Exploration & Understanding Rajarshi Guha School of Informatics Indiana University 16 th August, 2006 utline verview 1 verview 2 3 CDK R utline verview 1 verview 2 3 CDK
verview Chemical Space: Modeling Exploration & Understanding Rajarshi Guha School of Informatics Indiana University 16 th August, 2006 utline verview 1 verview 2 3 CDK R utline verview 1 verview 2 3 CDK
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Introduction to Machine Learning Midterm Exam
 10-701 Introduction to Machine Learning Midterm Exam Instructors: Eric Xing, Ziv Bar-Joseph 17 November, 2015 There are 11 questions, for a total of 100 points. This exam is open book, open notes, but
10-701 Introduction to Machine Learning Midterm Exam Instructors: Eric Xing, Ziv Bar-Joseph 17 November, 2015 There are 11 questions, for a total of 100 points. This exam is open book, open notes, but
Molecular Descriptors Theory and tips for real-world applications
 Molecular Descriptors Theory and tips for real-world applications Francesca Grisoni University of Milano-Bicocca, Dept. of Earth and Environmental Sciences, Milan, Italy ETH Zurich, Dept. of Chemistry
Molecular Descriptors Theory and tips for real-world applications Francesca Grisoni University of Milano-Bicocca, Dept. of Earth and Environmental Sciences, Milan, Italy ETH Zurich, Dept. of Chemistry
6.036 midterm review. Wednesday, March 18, 15
 6.036 midterm review 1 Topics covered supervised learning labels available unsupervised learning no labels available semi-supervised learning some labels available - what algorithms have you learned that
6.036 midterm review 1 Topics covered supervised learning labels available unsupervised learning no labels available semi-supervised learning some labels available - what algorithms have you learned that
Analysis of a Large Structure/Biological Activity. Data Set Using Recursive Partitioning and. Simulated Annealing
 Analysis of a Large Structure/Biological Activity Data Set Using Recursive Partitioning and Simulated Annealing Student: Ke Zhang MBMA Committee: Dr. Charles E. Smith (Chair) Dr. Jacqueline M. Hughes-Oliver
Analysis of a Large Structure/Biological Activity Data Set Using Recursive Partitioning and Simulated Annealing Student: Ke Zhang MBMA Committee: Dr. Charles E. Smith (Chair) Dr. Jacqueline M. Hughes-Oliver
MultiCASE CASE Ultra model for severe skin irritation in vivo
 MultiCASE CASE Ultra model for severe skin irritation in vivo 1. QSAR identifier 1.1 QSAR identifier (title) MultiCASE CASE Ultra model for severe skin irritation in vivo, Danish QSAR Group at DTU Food.
MultiCASE CASE Ultra model for severe skin irritation in vivo 1. QSAR identifier 1.1 QSAR identifier (title) MultiCASE CASE Ultra model for severe skin irritation in vivo, Danish QSAR Group at DTU Food.
Drug Design 2. Oliver Kohlbacher. Winter 2009/ QSAR Part 4: Selected Chapters
 Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Grouping and Read-Across for Respiratory Sensitisation. Dr Steve Enoch School of Pharmacy and Biomolecular Sciences Liverpool John Moores University
 Grouping and Read-Across for Respiratory Sensitisation Dr Steve Enoch School of Pharmacy and Biomolecular Sciences Liverpool John Moores University Chemicals are grouped into a category Toxicity data from
Grouping and Read-Across for Respiratory Sensitisation Dr Steve Enoch School of Pharmacy and Biomolecular Sciences Liverpool John Moores University Chemicals are grouped into a category Toxicity data from
Quiz QSAR QSAR. The Hammett Equation. Hammett s Standard Reference Reaction. Substituent Effects on Equilibria
 Quiz Select a method you are using for your project and write ~1/2 page discussing the method. Address: What does it do? How does it work? What assumptions are made? Are there particular situations in
Quiz Select a method you are using for your project and write ~1/2 page discussing the method. Address: What does it do? How does it work? What assumptions are made? Are there particular situations in