Direction dependent thermal conductivity of monolayer. phosphorene: parameterization of Stillinger-Weber potential and. molecular dynamics study
|
|
|
- Robyn Allen
- 6 years ago
- Views:
Transcription
1 Direction dependent thermal conductivity of monolayer phosphorene: parameterization of Stillinger-Weber potential and molecular dynamics study Wen Xu, 1, 2 Liyan Zhu, 3 Yongqing Cai, 2 Gang Zhang, 2, * 1, 4, 5 and Baowen Li 1 Department of Physics, Centre for Advanced 2D Material and Centre for Computational Science and Engineering, National University of Singapore, Singapore , Singapore 2 Institute of High Performance Computing, A*STAR, Singapore , Singapore 3 Department of physics, Huai an Normal University, Jiangsu,223000, China 4 NUS Graduate School for Integrative Sciences and Engineering, National University of Singapore, Kent Ridge , Singapore 5 Center for Phononics and Thermal Energy Science, School of Physics Science and Engineering, Tongji University, Shanghai, China * zhangg@ihpc.a-star.edu.sg Abstract A Stillinger-Weber interatomic potential is parameterized for phosphorene. It well reproduces the crystal structure, cohesive energy and phonon dispersion predicted by first-principles calculations. The thermal conductivity of phosphorene is further explored by equilibrium molecular dynamics simulations adopting the optimal set of potential parameters. At room temperature, the intrinsic thermal conductivities along zigzag and armchair directions are about and 33.0 W/mK, respectively, with a large anisotropy ratio of five. The remarkably directional dependence of thermal conductivity in phosphorene, consistent with previous reports, is mainly due to the strong anisotropy of phonon group velocities, and weak anisotropy of phonon lifetimes as revealed by lattice dynamics calculations. Moreover, the effective phonon mean free paths at zigzag and armchair directions are about and 43.4nm, respectively. 1
2 I. INTRODUCTION With the continuous downscaling of electronic devices, short channel effects have severely affected their performance. Fortunately, the two-dimensional(2d) semiconductors, such as MoS 2 [1], are immune to these effects, which makes 2D semiconducting nanosheets receive a significant amount of attention. Most recently, another novel 2D semiconductor, monolayer phosphorene, which exhibits fascinating physical properties including a sizable direct band gap, high carrier mobilities, and a large on-off current ratio[2, 3], has been exfoliated. These superior electronic properties render phosphorene a promising candidate in many nanoelectronic and optoelectronic applications. In addition to the electronic and optical properties, thermal properties of nano materials have also attracted considerable attention due to their unique features different from their counterpart in macroscale[4]. On one hand, thermal management in nanoscale devices with a high power density is a critical issue, where a high thermal conductivity material is needed to dissipate the Joule heat as quickly as possible. On the other hand, phosphorene has been predicted to be a potential thermoelectric material[5, 6], where a low thermal conductivity is desired to achieve a high efficiency. With either motivation, it is necessary to perform a thorough study on the thermal transport properties of phosphorene. Since phosphorene is a semiconductor, the dominant thermal carriers in it are phonons. So far, theoretical assessment of phonon transport in phosphorene has been done with nonequilibrium Green s function (NEGF) method[7] or by solving the Boltzmann transport equation (BTE)[8, 9], while little is related to molecular dynamics (MD) simulations. MD simulation is another powerful tool to handle many-body problems at the atomic level. It approaches the thermal transport problems without the thermodynamic-limit assumption, and naturally includes full anharmonicity in atomistic interactions. Besides predicting the thermal 2
3 conductivity, the MD simulation is also very efficient in studying problems such as interfacial thermal resistance[10], thermal rectification[11], mechanical properties[12] and fractural process[13] of nano materials. The applications of MD simulations have covered a wide range of research systems, such as liquids, clusters, and biomolecules[14]. In MD simulations, the force on each atom according to the force field is calculated to numerically solve the Newton's equations of motion. Although a fully quantum-mechanical treatment of the system s Hamiltonian is highly desirable, it can only be applied to the system with a small size due to the large computational load. Therefore, tremendous efforts have been devoted to developing empirical potential fields that can be applied to a large system with the sacrifice of exactness. However, so far there is no existing empirical interatomic potential for monolayer phosphorene yet. Therefore, it is the demand to study thermal transport in phosphorene, the power of MD simulations and the lack of empirical interatomic potential for phosphorene that inspire our work reported in this paper. In this work, we use quantities obtained from first-principles calculations to parameterize an empirical potential for phosphorene, which is a usual strategy and has been applied to investigate thermal transport in various materials[15-17]. Specifically, the well-established Stillinger-Weber (SW) potential[18] is used as the prototype for fitting, due to its simple form and wide applications[17, 19, 20]. The parameterized potential well reproduces the crystal structure, cohesive energy and phonon dispersion of phosphorene predicted by first-principles calculations. Subsequently, MD simulations are performed with this potential to study the thermal transport in phosphorene. Good agreement between results in this work and previously reported results is reached. This paper is organized in the following way: In Sec. II, the parameterization of SW potential is described and the final parameter set is reported. In Sec. III, the thermal conductivity of phosphorene is calculated with equilibrium MD simulations. In Sec. IV, the phonon properties such as group velocities, lifetimes and mean 3
4 free paths are evaluated. Finally, in Sec. V, deficiencies of the present SW potential and future developing directions are discussed, followed with a summary of this paper. II. PARAMETERIZATION OF POTENTIAL Phosphorene consists of a sheet of phosphorus atoms puckered along the so-called armchair direction, with four atoms in each unit cell (see Figure 1). Noticing that the phosphorus atoms occupy two planes parallel with XY plane, we enumerate them with P1 and P2 accordingly. There are two nonequivalent types of P-P bonds, one is parallel, and the other is unparallel with XY plane. Similarly, there are two types of bond angles. We distinguish the two types of bonds and bond angles with subscripts in and out (see Figure 1(b)). The SW potential is expected to properly describe interactions at these bonds and bond angles. It is worth mentioning that P1 and P2 are just used for specifying the interatomic interactions as will be shown. According to first-principles calculations[8], the lattice constants at X and Y directions are 3.301Å and 4.601Å respectively. The other geometrical quantities are as follows: l in = Å, l out = Å, in = , and out = The SW potential was initially proposed to describe interactions in solid and liquid forms of Si[18]. The potential function comprises a two-body term which describes the bond length and a three-body term which describes the bond angle, and it could be written as,,, 1 where denotes the bond length between atom i and atom j, and denotes the bond angle formed by and, with atom i at the vertex. Since GULP[21] is used as the fitting tool, we further write the two terms in Eq. (1) with the form applied in GULP, 1, 2,, cos cos, 3 4
5 where is the cutoff distance for the interaction between atom i and atom j, is the equilibrium value for. If a vector composed by the candidate parameters in SW potential as,,,, is assumed, the fitting strategy in GULP is to find that minimizes the objective function F, 4 with the Broyden-Fletcher-Goldfarb-Shanno(BFGS) algorithm[22]. is the ith observable quantity, such as lattice constant, cohesive energy, elastic constant and so on, provided by experiments or first-principles calculations. is the corresponding value calculated using the SW potential with parameter set. is the weighting factor, determined by the importance of the observable quantity. A lower dimension of is preferred for a simpler optimization, but lower dimension also means fewer degrees of freedom to fit the observable quantities. In the realization on phosphorene, we first clarify the undetermined SW parameters. As is mentioned, in phosphorene there are two types of bonds, P1-P1 and P1-P2, while the P2-P2 bond is equivalent with the P1-P1 bond. Considering the two-body interaction term (see Eq. (2)), there are undetermined parameters,,,, and. There are two types of bond angles, P1-P1-P1 (the first atom at the vertex) and P1-P1-P2 (equivalent with the P2- P2-P1 as marked in Figure 1(b)). Considering the three-body interaction term (see Eq. (3)), and are set with the equilibrium values as 96 and 104, respectively. is assumed for simplification. Thus there are three more undetermined parameters, and. The cutoff distances have not been treated as undetermined parameters in fitting, and we manually adjust them. is assumed according to the similar 5
6 length of the two types of bond, and we finally set 2.8 Å, which is close to the average of the first and second nearest neighbor distances in phosphorene. Next, a proper set of observable quantities is needed as the targets in fitting. What we use are lattice constants and fractional coordinates (they describe the crystal structure, which is also characterized by bond lengths and bond angles), cohesive energy E and phonon frequencies. The phonon frequencies contain much information related to the thermal and mechanical properties. It is worth mentioning that the geometrical quantities should be given large weighting factors since they are generally of primary consideration. Moreover, a severe variation of lattice constants greatly varies the dimension of the first Brillouin zone (BZ), as a result, the site of a particular phonon mode in the reciprocal space shifts undesirably, making the fitting of phonon frequencies problematic. In this work, the values of observable quantities are all taken from first-principles calculations[8]. Besides the lattice constants mentioned above, the cohesive energy -3.48eV/atom, and phonon frequencies from 11 k- points (132 modes in total) in the first BZ along Y--X ( included, see Figure 2) are used as observables. The SW parameters for Si[18] are used as the initial guess. The fitting is performed for several rounds, in each following round the initial parameter set is the result of last fitting with a slight modification. Thus far, the most satisfactory parameter set discovered by us is the following: eV, eV, Å, Å, Å, Å, eV, eV and Å. We can also translate them into the more general form as in the original SW potential[18], and the full result is listed in Table I in the style of potential files in LAMMPS[23]. The lattice constants at X and Y directions of the relaxed SW phosphorene are 3.284Å and 4.590Å, very close to the first principles results 3.301Å and 4.601Å, respectively. More comparisons are listed in Table II. The present SW potential gives a fairly well reproduction 6
7 of geometrical parameters and cohesive energy, while the acoustic velocities are generally underestimated by about 20%, which could actually be detected from the phonon dispersion comparison shown in Figure 2. In total, the SW potential parameter set developed in the present work gives a well reproduction of the acoustic phonon branches and most of the optical phonon branches, despite of deviation due to the oversimplification in this classical potential, but which is not a serious problem here if the dominant role of acoustic phonons in thermal transport is considered. Interestingly, in the armchair direction the transverse acoustic velocity is higher than the longitudinal one, which might be related to the specially puckered structure of phosphorene. III. THERMAL CODUCTIVITY CALCULATION Before applying the parameterized SW potential to calculate the thermal conductivity of phosphorene, it is necessary to test the reliability of it at nonzero temperatures, since the fitting is performed with reference at the ground state (T = 0K). The testing is done by thermalizing the SW phosphorene at a particular temperature and examining the structural stability. MD simulations are performed with LAMMPS[23]. We choose a phosphorene supercell of 1010 unit cells (UCs) as the testing sample, and the crystal direction is the same as in Figure 1(a), namely, the zigzag and armchair directions are along X and Y axes, respectively. Periodic boundary condition is applied to all directions, and thick enough vacuum layer (20Å) is adopted in the simulating box at the Z direction to avoid unwanted layer-layer interaction. The time step is set as 1.0fs in all the following MD runs. We thermalize the sample in NPT ensemble at 1000K for 10 6 time steps, where the strain (slight) in X and Y directions is released by varying the box size, and then run the simulation in NVE ensemble for another 10 6 steps. Nevertheless, amorphization arises if the atomic neighbor list within the cutoff distance is frequently updated. The puckered phosphorene is flexible, especially along the armchair direction. Thus, the non-bonded atoms (beyond the 7
8 first nearest neighbors) have many chances to be closer (within the potential s cutoff distance) in simulations, and amorphization arises if these atoms are treated as interaction pairs, since the SW potential tends to pull them into a wrong configuration. In fact, the similar amorphization incurred by unwanted neighbors also arises in Si described by the original SW potential, as what happened on the (001) surface of Si nanowire in MD simulations[24]. To avoid this problem, fixed atomic neighbor list is used (or only nearest neighbor interactions are considered). With this measure, the puckered structure of phosphorene is well maintained and no lattice distortion is found. It should be emphasized that the constrained neighbor list prevents the possibility in searching for other phosphorus allotropes, which should be one of the applications for MD simulation with empirical potentials. However, as a preliminary work, and our major concern is the thermal property of phosphorene, this treatment is an acceptable matter of expediency. To characterize the structural stability, the radial distribution function g(r) is evaluated (as shown in Figure 3(a)). The calculation of radial distribution function adopted here just considers atoms within the neighbor lists, but could effectively characterize the variation of bond lengths. The peak is well located and consistent with the equilibrium geometry of phosphorene. In addition, the time dependent kinetic energy and potential energy in the system are recorded in NVE ensemble (see Figure 3(b)). The equipartition theorem is approximately obeyed and the total energy is conserved. Next, the thermal conductivity of phosphorene is calculated with Green-Kubo formula[25]. We follow the expression in Ref. [26], κ 1 0, 5 where κ is the component of thermal conductivity tensor, is the Boltzmann constant, and are temperature and volume of the system, respectively. is the component of heat current at Cartesian direction (X, Y, and Z), and the angle brackets denote ensemble 8
9 average, equivalent to time average in the MD simulations. The integral part is called heat current autocorrelation function (HCAF). The heat current is defined as, 6 where is the time dependent coordinate of atom and is the site energy. It is calculated using LAMMPS. In its application on phosphorene, κ and κ correspond to κ and κ (subscript zig denotes zigzag and arm denotes armchair) respectively. With a phosphorene sample of UCs, periodic boundary condition is applied in all directions as in the potential testing procedure. MD simulation is first run in NVT ensemble for 10 6 steps, then switched to NVE ensemble, after another 10 6 steps, the heat current at each time step is dumped in the next steps. The HCAF at time 0,1,2 is evaluated as 0 1, 7 where is the time step and is total length of HCAF. With HCAF, Eq. (5) is used to calculate the thermal conductivity, where the integration is done by the trapezoidal rule, without any fitting. One typical calculation is shown in Figure 4. Nevertheless, it is not easy to specify the converged value in Green-Kubo method, since the integrated curve is fluctuating with thermal noise. To deal with this issue, for each data point, ten independent runs are performed and averaged, each with the same upper integration limit of 200ps. The Green-Kubo method is a formally exact approach[27] based on the fluctuationdissipation theorem. In principle, equilibrium MD calculation has the advantage that the sample size has no limit on phonons with comparable or larger mean free paths, thus there is no finite-size effect[26]. Nevertheless, usually, there is still size effect from two competing factors[27]. As the simulation sample gets larger, low frequency phonons near point, which 9
10 generally make significant contributions to thermal conductivity, are excited. On the other side, the possibility of phonon-phonon scattering, which tends to decreases the thermal conductivity, is enhanced. To eliminate this size effect, the simulating sample size is gradually increased from 1010 to 7070 UCs (the atoms are increased from 400 to 19600) and MD simulations are performed on each. The Green-Kubo method is formally exact only in the thermodynamic limit. Thus in equilibrium molecular dynamics simulations, periodic boundary conditions are adopted with a sufficiently large supercell size. As shown in Figure 5, it is clear that while κ shows little size dependence, κ gradually increases with the increase of super cell size and saturates to a constant value after N = 40. Thus the saturated value is treated as the thermal conductivity of ideal 2D phosphorene with N goes to infinity (thermodynamic limit). Interestingly, the in-plane thermal conductivity of phosphorene reveals strong anisotropy, the value at zigzag direction is four times larger than that at armchair direction, and this finding is consistent with other works[7-9]. The thermal conductivity of phosphorene over a wide range of temperatures is shown in Figure 6. The thermal conductivities at both directions (zigzag and armchair) gradually decrease with the increase of temperature. They roughly obey the law[8] but there is an obvious deviation when the temperature is high. Actually, the law could be deduced from the BTE approach under the following condition and assumption: when the temperature is high enough and when there is only cubic anharmonicity, the specific heat is almost temperature independent and the phonon scattering rates scale linearly with temperatures[28]. Nevertheless, with a full consideration of anharmonicity in MD simulations, the deviation from this law at high temperature is not surprising. In addition to the temperature dependence, the strong anisotropy (κ /κ 5.0) exists at all temperatures considered. Our calculation shows that the room temperature (T=300K) thermal conductivities of phosphorene at zigzag and armchair directions are and W/mK, 10
11 respectively. Recently, using DFT calculation with a full solution of the BTE, Jain and McGaughey reported the strongly anisotropic in-plane thermal conductivity in monolayer phosphorene. Their values are 110 and 36 W/mK for zigzag and armchair directions, respectively [9] Thus our MD results are in considerably good agreement with the previous work. The residual differences between our MD predicted thermal conductivities and their values could be due to one or more of the following reasons. First, in principle, each phonon mode is equally excited in classical MD simulations, which is different from the quantum statistical distribution in BTE method. Second, the SW potential could not exactly reproduce the phonon dispersion predicted by first-principles calculations, which directly influences the phonon group velocities and the phonon-phonon scattering rates. Third, in BTE calculations, the translational invariance and truncation at third-order force constants also significantly affect the calculated value of thermal conductivity [8, 9]. On the whole, the MD simulations provide fairly good agreement with first-principles based BTE results, and the most important characteristic in phosphorene, the strong anisotropic thermal conductivity, is well revealed by MD calculations with the parameterized empirical potential. IV. PHONON INFORMATION We have calculated the thermal conductivity of phosphorene. Nevertheless, the Green- Kubo method does not provide any mode-wise information, such as phonon group velocities, lifetimes and mean free paths (MFPs), which are commonly of interest. In this section, we use lattice dynamics calculations and MD simulations to evaluate these quantities in phosphorene, which are actually based on the relaxation time approximation (RTA). These quantities could explain the strong anisotropy of thermal conductivity in phosphorene. With the RTA, the thermal conductivity may be calculated as 11
12 ,, 8 where is the specific heat contributed by a phonon mode with wave vector at dispersion branch, equal to in the classical limit in MD simulations, is the component of group velocity at Cartesian direction, and is phonon lifetime (or relaxation time). For phosphorene, only those modes at -X and -Y (see Figure 2) in the first BZ are considered, since they are the most representative ones for zigzag and armchair directions, respectively. The dimension of sample used for phonon lifetime analysis is 4040, thus, the allowed wave vectors are 20,0,0 and 0, 20,0, where and are integers from 1 to 20 (half first BZ), and are lattice constants at zigzag and armchair directions, respectively. The phonon group velocities are calculated with a backward difference technique on the phonon dispersion calculated with GULP. The phonon lifetime is evaluated with frequency-domain normal mode decomposition[29] as following. We project the atomic velocities into reciprocal space as ;, ;, 9 where is normal mode velocity, and ; are the equilibrium position and time dependent velocity of the th atom in the th UC, is the complex conjugate of the eigenvector associated with the th atom at a given mode, is atomic mass and is the total number of UCs. Then the power spectrum of ; is evaluated with Fourier transformation, Φ, ; 0 ;
13 A Lorentz fitting of Φ, is performed in the neighborhood of the harmonic eigen frequency as Φ C, 4, 11 where C, and are unknown parameters. is the anharmonic eigen frequency at non-zero temperature, usually with a small shift from. The phonon lifetime is equal to 1 2, inverse to the width of the Lorentz peak. The atomic velocities are recorded every 20 time steps (0.02ps) in the NVE ensemble at 300K, and six independent runs are performed to minimize the uncertainty. One typical calculation is illustrated in Figure 7. The frequency dependent phonon group velocities and lifetimes are shown in Figure 8. Generally, the phonon group velocities at zigzag direction (-X) are higher than those of phonons at armchair (-Y) direction with similar frequencies, this is especially obvious for acoustic phonons, which make dominant contributions to the thermal transport as already proved[7-9]. In contrast, phonon lifetimes at the two directions are very close to each other except that the lifetimes at zigzag direction are a little longer than those in the armchair direction within 2~5THz and the optical phonon part. Therefore, according to Eq. (8), we speculate that the remarkably directional dependence of thermal conductivity is mainly due to the anisotropy of phonon group velocities. A rough estimation with the acoustic velocity ratio (about two) and Eq. (8) reveals a thermal conductivity ratio of four at the two directions, which is close to the ratio of five predicted by Green-Kubo method. We could evaluate the mode dependent phonon MFP as. Whereas, an effective MFP is usually of more interest since it is useful when designing the functional 13
14 nanostructures. For instance, it could be used to judge at which length scale the wave effect of phonons is important, so that the phononic crystal[30] with a proper period length efficiently works. The effective MFP is defined as [31]: (12) where κ is the intrinsic thermal conductivity of material at diffusive transport region, is the thermal conductance at the ballistic transport limit, and is the so called effective MFP. Using first-principles calculations and NEGF method, thermal conductance and MFP of MoS 2 sheet [31] have been studied, and this formula has been used to evaluate the value of thermal conductivity. To evaluate of phosphorene, we make use of the intrinsic thermal conductivity κ from Green-Kubo method in this work and the ballistic thermal conductance in our previous work[7]. At 300K, W/mK, 33.0 W/m, W/m K and W/m K. Thus, the effective MFPs are about and 43.4nm at zigzag and armchair directions, respectively. The MFPs of phosphorene are one order of magnitude lower than that of graphene[32], while one order higher than that of MoS 2 [33]. V. DSICUSSION AND SUMMARY Although satisfying descriptions of the crystal structure, cohesive energy, phonon dispersion and thermal conductivity of monolayer black phosphorene are reproduced, it must be mentioned that this is a preliminary work on phosphorene from such an approach, despite of some deficiencies. The most significant limitation is related to the reliability of the set of potential parameters fitted in the present work. First, this interatomic potential is specified for black phosphorene. The enumeration of phosphorous atoms, bond lengths and bond angles (see Table I) may limit its application to other phosphorus allotropes such as the blue 14
15 phosphorus [34]. A more widely transferable empirical interatomic potential might call for a more complex form, such as the Tersoff [35], Brenner [36] and EDIP [37] types. Besides, a long range force between non-bonded atoms might also need to be considered. Furthermore, the fixed atomic neighbor list adopted in our calculation influences other possible applications, such as modeling melting, deformation and fracture. The amorphization phenomenon predicted by this potential when all neighbor pairs are treated shows that the underlying description may be incomplete. All of these show that further investigations are deserved with this potential as a starting point. In summary, we have parameterized a set of Stilling-Weber empirical potential parameters for monolayer black phosphorene, and evaluated the thermal conductivity by using equilibrium molecular dynamics simulations. Unlike the isotropic in-plane thermal conductivity in graphene and MoS 2, thermal transport in monolayer black phosphorene exists a strongly directional dependence, with the values of room temperature thermal conductivity along zigzag and armchair directions are and 33.0 W/mK, respectively. The MD predicted values are close with results from first-principles and BTE calculations. 15
16 References [1] H. Liu, A. T. Neal, and P. D. Ye, ACS nano 6, 8563 (2012). [2] L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen, and Y. Zhang, Nat Nano 9, 372 (2014). [3] H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tománek, and P. D. Ye, ACS Nano 8, 4033 (2014). [4] N. Yang, X. Xu, G. Zhang, and B. Li, AIP Advances 2, (2012). [5] R. Fei, A. Faghaninia, R. Soklaski, J.-A. Yan, C. Lo, and L. Yang, Nano Letters 14, 6393 (2014). [6] J. Zhang, H. J. Liu, L. Cheng, J. Wei, J. H. Liang, D. D. Fan, J. Shi, X. F. Tang, and Q. J. Zhang, Sci. Rep. 4 (2014). [7] Z.-Y. Ong, Y. Cai, G. Zhang, and Y.-W. Zhang, The Journal of Physical Chemistry C 118, (2014). [8] L. Zhu, G. Zhang, and B. Li, Physical Review B 90, (2014). [9] A. Jain and A. J. H. McGaughey, Sci. Rep. 5, 8501 (2015). [10] W. Xu, G. Zhang, and B. Li, Journal of Applied Physics 116, (2014). [11] R. Rurali, X. Cartoixà, and L. Colombo, Physical Review B 90, (2014). [12] N.-N. Li, Z.-D. Sha, Q.-X. Pei, and Y.-W. Zhang, The Journal of Physical Chemistry C 118, (2014). [13] Z. D. Sha, S. S. Quek, Q. X. Pei, Z. S. Liu, T. J. Wang, V. B. Shenoy, and Y. W. Zhang, Sci. Rep. 4, 5991 (2014). [14] G. Zhang, Nanoscale Energy Transport and Harvesting: A Computational Study (Pan Stanford, 2014). [15] B. Qiu and X. Ruan, Physical Review B 80, (2009). [16] C. Sevik, A. Kinaci, J. B. Haskins, and T. Çağın, Physical Review B 84, (2011). [17] X. Zhang, H. Xie, M. Hu, H. Bao, S. Yue, G. Qin, and G. Su, Physical Review B 89, (2014). [18] F. H. Stillinger and T. A. Weber, Physical Review B 31, 5262 (1985). [19] X. W. Zhou, D. K. Ward, J. E. Martin, F. B. van Swol, J. L. Cruz-Campa, and D. Zubia, Physical Review B 88, (2013). [20] J. Chen, G. Zhang, and B. Li, The Journal of Chemical Physics 135, (2011). [21] J. D. Gale and A. L. Rohl, Molecular Simulation 29, 291 (2003). [22] D. F. Shanno, Mathematics of computation 24, 647 (1970). [23] S. Plimpton, Journal of computational physics 117, 1 (1995). [24] S. G. Volz and G. Chen, Applied Physics Letters 75, 2056 (1999). [25] R. Kubo, Journal of the Physical Society of Japan 12, 570 (1957). [26] P. K. Schelling, S. R. Phillpot, and P. Keblinski, Physical Review B 65, (2002). [27] A. J. Ladd, B. Moran, and W. G. Hoover, Physical Review B 34, 5058 (1986). [28] S. Bickham and J. Feldman, Physical Review B 57, (1998). [29] A. J. H. McGaughey and J. M. Larkin, Annual Review of Heat Transfer 17, 49 (2014). [30] L. Yang, N. Yang, and B. Li, Nano letters 14, 1734 (2014). [31] Y. Cai, J. Lan, G. Zhang, and Y.-W. Zhang, Physical Review B 89, (2014). [32] S. Ghosh, W. Bao, D. L. Nika, S. Subrina, E. P. Pokatilov, C. N. Lau, and A. A. Balandin, Nat Mater 9, 555 (2010). [33] X. Liu, G. Zhang, Q.-X. Pei, and Y.-W. Zhang, Applied Physics Letters 103, (2013). [34] Z. Zhu and D. Tománek, Phys. Rev. Lett. 112, (2014). [35] J. Tersoff, Physical Review B 39, 5566 (1989). [36] D. W. Brenner, Physical Review B 42, 9458 (1990). [37] M. Z. Bazant and E. Kaxiras, Physical Review Letters 77, 4370 (1996). 16
17 Table I. SW potential parameters for phosphorene. (ev) (Å) a A B p q tol P1P1P P2P2P P1P2P P2P1P P1P1P P1P2P P2P1P P2P2P Table II. Geometrical parameters, cohesive energy, and acoustic velocities predicted by firstprinciples calculations as compared to those predicted by SW potential. Values with * are cited from Ref. [34]. l in (Å) l out (Å) in () out () E (ev/atom) (Km/s) (Km/s) (Km/s) (Km/s) 1 st principles (7.8 * ) (3.8 * ) SW potential

















18 Figure 1. Schematic of phosphorene. (a) Top view of the 33 supercell, the red dashed lines indicate the unit cells, the zigzag and armchair directions are arranged along X and Y axes respectively. (b) Enlarged drawing of one unit cell (including full boundary atoms),, the top three (dark-coloured) and bottom three (light-coloured) phosphorus atomss are enumerated as P1 and P2 respectively. 18














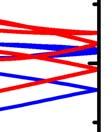


19 Figure 2 Phonon dispersions of phosphorene along Y--X in the t first BZ. The blue and red curves are obtained by first-principles calculations and the parameterized SW potential respectively. 19






20 Figure 3. SW phosphorene at 1000K. (a) Radial distribution function,, P1-P1 means the statistics of P1 atoms around P1 atoms, and P1-P2 means statistics of P22 atoms around P1 atoms. (b) The time dependent kinetic energy E k, potential energy E p, and half of total energy E tot of the sample, E p and E tot have been subtracted with their ground state values (when T=0K). 20








 The")
21 Figure 4. Evaluating thermal conductivity y of phosphorene at 300K with Green-Kubo method, the simulating sample is of 6060 UCs. The green dashed linee indicates the integration time limit. (a) The HCAF. (b) The dependencee of integration result on o the upperr limit. 21



22 Figure 5. Thermal conductivity of phosphorene at 300K calculated with samples of different sizes ( UCs) ). 22







23 Figure 6. Temperature dependent thermal conductivity of phosphorene. Values of and are scaled on the left and right vertical axes, respectively, and they have a ratio of five. The green dashed line denotess an extrapolation according to thee law. 23

")







,")


24 Figure 7. Power spectrum (blue dots) of a transverse acoustic phonon mode, whose wave vector and harmonic eigen frequency are,, and 2.998THz, respectively, and the Lorentz fitting (red curves), which shows that the eigen frequency is shifted to 2.955THz at 300K. The phononn lifetime is evaluated to be 31.7ps. 24















25 Figure 8. Phonon group velocities and lifetimes at zigzag and armchair directions. (a) Phonon group velocities. (b) Phonon lifetimes. 25
Molecular Dynamics Study of Thermal Rectification in Graphene Nanoribbons
 Molecular Dynamics Study of Thermal Rectification in Graphene Nanoribbons Jiuning Hu 1* Xiulin Ruan 2 Yong P. Chen 3# 1School of Electrical and Computer Engineering and Birck Nanotechnology Center, Purdue
Molecular Dynamics Study of Thermal Rectification in Graphene Nanoribbons Jiuning Hu 1* Xiulin Ruan 2 Yong P. Chen 3# 1School of Electrical and Computer Engineering and Birck Nanotechnology Center, Purdue
T wo-dimensional (2D) materials (e.g., graphene, MoS2, silicene) are a focus of intense research because of
 materials (e.g., graphene, MoS2, silicene) are a focus of intense research because of") OPEN SUBJECT AREAS: TWO-DIMENSIONAL MATERIALS APPLIED PHYSICS Strongly anisotropic in-plane thermal transport in single-layer black phosphorene Ankit Jain & Alan J. H. McGaughey Received 23 October 2014
OPEN SUBJECT AREAS: TWO-DIMENSIONAL MATERIALS APPLIED PHYSICS Strongly anisotropic in-plane thermal transport in single-layer black phosphorene Ankit Jain & Alan J. H. McGaughey Received 23 October 2014
Molecular Dynamics Study of Thermal Rectification in Graphene Nanoribbons
 Int J Thermophys (2012) 33:986 991 DOI 10.1007/s10765-012-1216-y Molecular Dynamics Study of Thermal Rectification in Graphene Nanoribbons Jiuning Hu Xiulin Ruan Yong P. Chen Received: 26 June 2009 / Accepted:
Int J Thermophys (2012) 33:986 991 DOI 10.1007/s10765-012-1216-y Molecular Dynamics Study of Thermal Rectification in Graphene Nanoribbons Jiuning Hu Xiulin Ruan Yong P. Chen Received: 26 June 2009 / Accepted:
Carbon Nanocone: A Promising Thermal Rectifier
 Carbon Nanocone: A Promising Thermal Rectifier Nuo Yang 1, Gang Zhang 2, a) 3,1, b) and Baowen Li 1 Department of Physics and Centre for Computational Science and Engineering, National University of Singapore,
Carbon Nanocone: A Promising Thermal Rectifier Nuo Yang 1, Gang Zhang 2, a) 3,1, b) and Baowen Li 1 Department of Physics and Centre for Computational Science and Engineering, National University of Singapore,
Supporting Information
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2015 Supporting Information Single Layer Lead Iodide: Computational Exploration of Structural, Electronic
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2015 Supporting Information Single Layer Lead Iodide: Computational Exploration of Structural, Electronic
Tinselenidene: a Two-dimensional Auxetic Material with Ultralow Lattice Thermal Conductivity and Ultrahigh Hole Mobility
 Tinselenidene: a Two-dimensional Auxetic Material with Ultralow Lattice Thermal Conductivity and Ultrahigh Hole Mobility Li-Chuan Zhang, Guangzhao Qin, Wu-Zhang Fang, Hui-Juan Cui, Qing-Rong Zheng, Qing-Bo
Tinselenidene: a Two-dimensional Auxetic Material with Ultralow Lattice Thermal Conductivity and Ultrahigh Hole Mobility Li-Chuan Zhang, Guangzhao Qin, Wu-Zhang Fang, Hui-Juan Cui, Qing-Rong Zheng, Qing-Bo
The Vacancy Effect on Thermal Interface Resistance between Aluminum and Silicon by Molecular Dynamics
 The Vacancy Effect on Thermal Interface Resistance between Aluminum and Silicon by Molecular Dynamics Journal: 2014 MRS Fall Meeting Manuscript ID: 2035346.R1 Manuscript Type: Symposium NN Date Submitted
The Vacancy Effect on Thermal Interface Resistance between Aluminum and Silicon by Molecular Dynamics Journal: 2014 MRS Fall Meeting Manuscript ID: 2035346.R1 Manuscript Type: Symposium NN Date Submitted
Bandgap Modulated by Electronic Superlattice in Blue Phosphorene
 Bandgap Modulated by Electronic Superlattice in Blue Phosphorene Jincheng Zhuang,,,# Chen Liu,,# Qian Gao, Yani Liu,, Haifeng Feng,, Xun Xu, Jiaou Wang, Shi Xue Dou,, Zhenpeng Hu, and Yi Du*,, Institute
Bandgap Modulated by Electronic Superlattice in Blue Phosphorene Jincheng Zhuang,,,# Chen Liu,,# Qian Gao, Yani Liu,, Haifeng Feng,, Xun Xu, Jiaou Wang, Shi Xue Dou,, Zhenpeng Hu, and Yi Du*,, Institute
arxiv:cond-mat/ v3 [cond-mat.mes-hall] 2 Nov 2005
![arxiv:cond-mat/ v3 [cond-mat.mes-hall] 2 Nov 2005](/thumbs/89/100586496.jpg "arxiv:cond-mat/ v3 [cond-mat.mes-hall] 2 Nov 2005") Thermal Conductivity of Nanotubes Revisited: Effects of Chirality, Isotope Impurity, Tube Length, and Temperature arxiv:cond-mat/0403393v3 [cond-mat.mes-hall] 2 Nov 2005 Gang Zhang Department of Physics,
Thermal Conductivity of Nanotubes Revisited: Effects of Chirality, Isotope Impurity, Tube Length, and Temperature arxiv:cond-mat/0403393v3 [cond-mat.mes-hall] 2 Nov 2005 Gang Zhang Department of Physics,
Hydrogenation of Penta-Graphene Leads to Unexpected Large. Improvement in Thermal Conductivity
 Supplementary information for Hydrogenation of Penta-Graphene Leads to Unexpected Large Improvement in Thermal Conductivity Xufei Wu, a Vikas Varshney, b,c Jonghoon Lee, b,c Teng Zhang, a Jennifer L. Wohlwend,
Supplementary information for Hydrogenation of Penta-Graphene Leads to Unexpected Large Improvement in Thermal Conductivity Xufei Wu, a Vikas Varshney, b,c Jonghoon Lee, b,c Teng Zhang, a Jennifer L. Wohlwend,
Phonon Coherent Resonance and Its Effect on Thermal Transport In. Core-Shell Nanowires
 Phonon Coherent Resonance and Its Effect on Thermal Transport In Core-Shell Nanowires Jie Chen, 1 Gang Zhang, 2, 1, 3, and Baowen Li 1 Department of Physics and Centre for Computational Science and Engineering,
Phonon Coherent Resonance and Its Effect on Thermal Transport In Core-Shell Nanowires Jie Chen, 1 Gang Zhang, 2, 1, 3, and Baowen Li 1 Department of Physics and Centre for Computational Science and Engineering,
SUPPLEMENTARY INFORMATION
 Thermal Conductivity of Isotopically Modified Graphene Shanshan Chen 1,, Qingzhi Wu, Columbia Mishra, Junyong Kang 1, Hengji Zhang 3 Kyeongjae Cho 3, Weiwei Cai 1, *, Alexander A. Balandin 4* and Rodney
Thermal Conductivity of Isotopically Modified Graphene Shanshan Chen 1,, Qingzhi Wu, Columbia Mishra, Junyong Kang 1, Hengji Zhang 3 Kyeongjae Cho 3, Weiwei Cai 1, *, Alexander A. Balandin 4* and Rodney
Size-dependent model for thin film and nanowire thermal conductivity
 AIP/23-QED Size-dependent model for thin film and nanowire thermal conductivity Alan J. H. McGaughey,, a) Eric S. Landry,, 2 Daniel P. Sellan, 3 and Cristina H. Amon, 3 ) Department of Mechanical Engineering,
AIP/23-QED Size-dependent model for thin film and nanowire thermal conductivity Alan J. H. McGaughey,, a) Eric S. Landry,, 2 Daniel P. Sellan, 3 and Cristina H. Amon, 3 ) Department of Mechanical Engineering,
Supplementary Figures
 Supplementary Figures 8 6 Energy (ev 4 2 2 4 Γ M K Γ Supplementary Figure : Energy bands of antimonene along a high-symmetry path in the Brillouin zone, including spin-orbit coupling effects. Empty circles
Supplementary Figures 8 6 Energy (ev 4 2 2 4 Γ M K Γ Supplementary Figure : Energy bands of antimonene along a high-symmetry path in the Brillouin zone, including spin-orbit coupling effects. Empty circles
arxiv: v2 [cond-mat.mtrl-sci] 24 Dec 2014
![arxiv: v2 [cond-mat.mtrl-sci] 24 Dec 2014](/thumbs/93/113232647.jpg "arxiv: v2 [cond-mat.mtrl-sci] 24 Dec 2014") Defect in Phosphorene arxiv:1411.6986v2 [cond-mat.mtrl-sci] 24 Dec 2014 Wei Hu 1, 2, 3, and Jinlong Yang 1 Computational Research Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
Defect in Phosphorene arxiv:1411.6986v2 [cond-mat.mtrl-sci] 24 Dec 2014 Wei Hu 1, 2, 3, and Jinlong Yang 1 Computational Research Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
Table of Contents. Table of Contents Using the Crystal Builder. Introduction Crystal structure of black phosphorus Phosphorene and its bandstructure
 Table of Contents Table of Contents Using the Crystal Builder Introduction Crystal structure of black phosphorus Phosphorene and its bandstructure Bandstructure References 1 2 2 3 7 9 10 QuantumATK Try
Table of Contents Table of Contents Using the Crystal Builder Introduction Crystal structure of black phosphorus Phosphorene and its bandstructure Bandstructure References 1 2 2 3 7 9 10 QuantumATK Try
arxiv: v1 [cond-mat.mtrl-sci] 6 Jul 2017
![arxiv: v1 [cond-mat.mtrl-sci] 6 Jul 2017](/thumbs/83/87839175.jpg "arxiv: v1 [cond-mat.mtrl-sci] 6 Jul 2017") Lower lattice thermal conductivity in than or monolayer: a first-principles study San-Dong Guo School of Physics, China University of Mining and Technology, Xuzhou 22111, Jiangsu, China arxiv:177.1752v1
Lower lattice thermal conductivity in than or monolayer: a first-principles study San-Dong Guo School of Physics, China University of Mining and Technology, Xuzhou 22111, Jiangsu, China arxiv:177.1752v1
Research Article Effect of Strain on Thermal Conductivity of Si Thin Films
 Nanomaterials Volume 2016, Article ID 4984230, 5 pages http://dx.doi.org/10.1155/2016/4984230 Research Article Effect of Strain on Thermal Conductivity of Si Thin Films Xingli Zhang 1 and Guoqiang Wu 2
Nanomaterials Volume 2016, Article ID 4984230, 5 pages http://dx.doi.org/10.1155/2016/4984230 Research Article Effect of Strain on Thermal Conductivity of Si Thin Films Xingli Zhang 1 and Guoqiang Wu 2
Reduction of Thermal Conductivity by Nanoscale 3D Phononic Crystal
 Page 1 Lina Yang 1, Nuo Yang 2a, Baowen Li 1,2b 1 Department of Physics and Centre for Computational Science and Engineering, National University of Singapore, Singapore 117542, Republic of Singapore 2
Page 1 Lina Yang 1, Nuo Yang 2a, Baowen Li 1,2b 1 Department of Physics and Centre for Computational Science and Engineering, National University of Singapore, Singapore 117542, Republic of Singapore 2
Strength and Stability Analysis of a Single Walled Black Phosphorus Tube under Axial Compression
 Strength and Stability Analysis of a Single Walled Black Phosphorus Tube under Axial Compression Kun Cai 1, 2, Jing Wan 1, Ning Wei 1, Qinghua Qin 2* 1 College of Water Resources and Architectural Engineering,
Strength and Stability Analysis of a Single Walled Black Phosphorus Tube under Axial Compression Kun Cai 1, 2, Jing Wan 1, Ning Wei 1, Qinghua Qin 2* 1 College of Water Resources and Architectural Engineering,
GRAPHENE BASED HIGH-PERFORMANCE THERMAL INTERFACE MATERIALS
 Proceedings of the Asian Conference on Thermal Sciences 2017, 1st ACTS March 26-30, 2017, Jeju Island, Korea ACTS-00064 GRAPHENE BASED HIGH-PERFORMANCE THERMAL INTERFACE MATERIALS Jie Chen 1 * 1 Center
Proceedings of the Asian Conference on Thermal Sciences 2017, 1st ACTS March 26-30, 2017, Jeju Island, Korea ACTS-00064 GRAPHENE BASED HIGH-PERFORMANCE THERMAL INTERFACE MATERIALS Jie Chen 1 * 1 Center
PHONON THERMAL PROPERTIES OF GRAPHENE ON HEXAGONAL BORON NITRIDE
 Proceedings of the Asian Conference on Thermal Sciences 217, 1st ACTS March 26-3, 217, Jeju Island, Korea PHONON THERMAL PROPERTIES OF GRAPHENE ON ACTS-P146 HEXAGONAL BORON NITRIDE Ji-Hang Zou, Bing-Yang
Proceedings of the Asian Conference on Thermal Sciences 217, 1st ACTS March 26-3, 217, Jeju Island, Korea PHONON THERMAL PROPERTIES OF GRAPHENE ON ACTS-P146 HEXAGONAL BORON NITRIDE Ji-Hang Zou, Bing-Yang
Violation of Fourier s law and anomalous heat diffusion in silicon nanowires
 Nano Today (2010) 5, 85 90 available at www.sciencedirect.com journal homepage: www.elsevier.com/locate/nanotoday RAPID COMMUNICATION Violation of Fourier s law and anomalous heat diffusion in silicon
Nano Today (2010) 5, 85 90 available at www.sciencedirect.com journal homepage: www.elsevier.com/locate/nanotoday RAPID COMMUNICATION Violation of Fourier s law and anomalous heat diffusion in silicon
A Universal Gauge for Thermal Conductivity of Silicon Nanowires. With Different Cross Sectional Geometries
 A Universal Gauge for Thermal Conductivity of Silicon Nanowires With Different Cross Sectional Geometries Jie Chen, 1 Gang Zhang, 2, and Baowen Li 1, 3 1 Department of Physics and Centre for Computational
A Universal Gauge for Thermal Conductivity of Silicon Nanowires With Different Cross Sectional Geometries Jie Chen, 1 Gang Zhang, 2, and Baowen Li 1, 3 1 Department of Physics and Centre for Computational
arxiv: v2 [cond-mat.mtrl-sci] 10 Jul 2018
![arxiv: v2 [cond-mat.mtrl-sci] 10 Jul 2018](/thumbs/81/84557778.jpg "arxiv: v2 [cond-mat.mtrl-sci] 10 Jul 2018") Linear response phonon dynamics of anisotropic black phosphorous monolayer: PAW mediated ab initio DFPT calculations Sushant Kumar Behera and Pritam Deb Advanced Functional Material Laboratory, Department
Linear response phonon dynamics of anisotropic black phosphorous monolayer: PAW mediated ab initio DFPT calculations Sushant Kumar Behera and Pritam Deb Advanced Functional Material Laboratory, Department
Multiscale modeling of heat conduction in graphene laminates
 Multiscale modeling of heat conduction in graphene laminates Bohayra Mortazavi * and Timon Rabczuk Institute of Structural Mechanics, Bauhaus-Universität Weimar, Marienstr. 15, D-99423 Weimar, Germany
Multiscale modeling of heat conduction in graphene laminates Bohayra Mortazavi * and Timon Rabczuk Institute of Structural Mechanics, Bauhaus-Universität Weimar, Marienstr. 15, D-99423 Weimar, Germany
Molecular Dynamics Simulation of Fracture of Graphene
 Molecular Dynamics Simulation of Fracture of Graphene Dewapriya M. A. N. 1, Rajapakse R. K. N. D. 1,*, Srikantha Phani A. 2 1 School of Engineering Science, Simon Fraser University, Burnaby, BC, Canada
Molecular Dynamics Simulation of Fracture of Graphene Dewapriya M. A. N. 1, Rajapakse R. K. N. D. 1,*, Srikantha Phani A. 2 1 School of Engineering Science, Simon Fraser University, Burnaby, BC, Canada
Characterize Individual Phonon Mode Contribution Directly from. Non-equilibrium Molecular Dynamics Simulation
 Characterize Individual Phonon Mode Contribution Directly from Non-equilibrium Molecular Dynamics Simulation Yanguang Zhou 1, Xiaoliang Zhang 2 1, 2, *, and Ming Hu 1 Aachen Institute for Advanced Study
Characterize Individual Phonon Mode Contribution Directly from Non-equilibrium Molecular Dynamics Simulation Yanguang Zhou 1, Xiaoliang Zhang 2 1, 2, *, and Ming Hu 1 Aachen Institute for Advanced Study
Electro-Thermal Transport in Silicon and Carbon Nanotube Devices E. Pop, D. Mann, J. Rowlette, K. Goodson and H. Dai
 Electro-Thermal Transport in Silicon and Carbon Nanotube Devices E. Pop, D. Mann, J. Rowlette, K. Goodson and H. Dai E. Pop, 1,2 D. Mann, 1 J. Rowlette, 2 K. Goodson 2 and H. Dai 1 Dept. of 1 Chemistry
Electro-Thermal Transport in Silicon and Carbon Nanotube Devices E. Pop, D. Mann, J. Rowlette, K. Goodson and H. Dai E. Pop, 1,2 D. Mann, 1 J. Rowlette, 2 K. Goodson 2 and H. Dai 1 Dept. of 1 Chemistry
Electronic Supplementary Information
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2016 Electronic Supplementary Information Two-dimensional BX (X=P, As, Sb) Semiconductors with Mobilities
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2016 Electronic Supplementary Information Two-dimensional BX (X=P, As, Sb) Semiconductors with Mobilities
STRUCTURAL AND MECHANICAL PROPERTIES OF AMORPHOUS SILICON: AB-INITIO AND CLASSICAL MOLECULAR DYNAMICS STUDY
 STRUCTURAL AND MECHANICAL PROPERTIES OF AMORPHOUS SILICON: AB-INITIO AND CLASSICAL MOLECULAR DYNAMICS STUDY S. Hara, T. Kumagai, S. Izumi and S. Sakai Department of mechanical engineering, University of
STRUCTURAL AND MECHANICAL PROPERTIES OF AMORPHOUS SILICON: AB-INITIO AND CLASSICAL MOLECULAR DYNAMICS STUDY S. Hara, T. Kumagai, S. Izumi and S. Sakai Department of mechanical engineering, University of
University of Chinese Academy of Sciences, Beijing , People s Republic of China,
 SiC 2 Siligraphene and Nanotubes: Novel Donor Materials in Excitonic Solar Cell Liu-Jiang Zhou,, Yong-Fan Zhang, Li-Ming Wu *, State Key Laboratory of Structural Chemistry, Fujian Institute of Research
SiC 2 Siligraphene and Nanotubes: Novel Donor Materials in Excitonic Solar Cell Liu-Jiang Zhou,, Yong-Fan Zhang, Li-Ming Wu *, State Key Laboratory of Structural Chemistry, Fujian Institute of Research
Modeling thermal conductivity: a Green-Kubo approach
 Modeling thermal conductivity: a Green-Kubo approach Fabiano Oyafuso, Paul von Allmen, Markus Bühler Jet Propulsion Laboratory Pasadena, CA Funding: DARPA Outline Motivation -- thermoelectrics Theory Implementation
Modeling thermal conductivity: a Green-Kubo approach Fabiano Oyafuso, Paul von Allmen, Markus Bühler Jet Propulsion Laboratory Pasadena, CA Funding: DARPA Outline Motivation -- thermoelectrics Theory Implementation
FUNDAMENTAL ISSUES IN NANOSCALE HEAT TRANSFER: FROM COHERENCE TO INTERFACIAL RESISTANCE IN HEAT CONDUCTION
 HEFAT2014 10 th International Conference on Heat Transfer, Fluid Mechanics and Thermodynamics 14 16 July 2014 Orlando, Florida FUNDAMENTAL ISSUES IN NANOSCALE HEAT TRANSFER: FROM COHERENCE TO INTERFACIAL
HEFAT2014 10 th International Conference on Heat Transfer, Fluid Mechanics and Thermodynamics 14 16 July 2014 Orlando, Florida FUNDAMENTAL ISSUES IN NANOSCALE HEAT TRANSFER: FROM COHERENCE TO INTERFACIAL
Supporting Information
 Copyright WILEY-VCH Verlag GmbH & Co. KGaA, 69469 Weinheim, Germany, 2015. Supporting Information for Adv. Funct. Mater., DOI: 10.1002/adfm.201503131 Tuning the Excitonic States in MoS 2 /Graphene van
Copyright WILEY-VCH Verlag GmbH & Co. KGaA, 69469 Weinheim, Germany, 2015. Supporting Information for Adv. Funct. Mater., DOI: 10.1002/adfm.201503131 Tuning the Excitonic States in MoS 2 /Graphene van
VIRTUAL LATTICE TECHNIQUE AND THE INTERATOMIC POTENTIALS OF ZINC-BLEND-TYPE BINARY COMPOUNDS
 Modern Physics Letters B, Vol. 16, Nos. 5 & 6 (2002) 187 194 c World Scientific Publishing Company VIRTUAL LATTICE TECHNIQUE AND THE INTERATOMIC POTENTIALS OF ZINC-BLEND-TYPE BINARY COMPOUNDS LIU YING,
Modern Physics Letters B, Vol. 16, Nos. 5 & 6 (2002) 187 194 c World Scientific Publishing Company VIRTUAL LATTICE TECHNIQUE AND THE INTERATOMIC POTENTIALS OF ZINC-BLEND-TYPE BINARY COMPOUNDS LIU YING,
Thermal transport from first-principles DFT calculations. Keivan Esfarjani MIT. Department of Mechanical Engineering. 5/23/2012 Phonon UWM 1
 Thermal transport from first-principles DFT calculations Keivan Esfarjani Department of Mechanical Engineering MIT 5/23/2012 Phonon School @ UWM 1 Classical MD simulations use an empirical potential fitted
Thermal transport from first-principles DFT calculations Keivan Esfarjani Department of Mechanical Engineering MIT 5/23/2012 Phonon School @ UWM 1 Classical MD simulations use an empirical potential fitted
Thermal Transport in Graphene and other Two-Dimensional Systems. Li Shi. Department of Mechanical Engineering & Texas Materials Institute
 Thermal Transport in Graphene and other Two-Dimensional Systems Li Shi Department of Mechanical Engineering & Texas Materials Institute Outline Thermal Transport Theories and Simulations of Graphene Raman
Thermal Transport in Graphene and other Two-Dimensional Systems Li Shi Department of Mechanical Engineering & Texas Materials Institute Outline Thermal Transport Theories and Simulations of Graphene Raman
Spin Lifetime Enhancement by Shear Strain in Thin Silicon-on-Insulator Films. Dmitry Osintsev, Viktor Sverdlov, and Siegfried Selberherr
 10.1149/05305.0203ecst The Electrochemical Society Spin Lifetime Enhancement by Shear Strain in Thin Silicon-on-Insulator Films Dmitry Osintsev, Viktor Sverdlov, and Siegfried Selberherr Institute for
10.1149/05305.0203ecst The Electrochemical Society Spin Lifetime Enhancement by Shear Strain in Thin Silicon-on-Insulator Films Dmitry Osintsev, Viktor Sverdlov, and Siegfried Selberherr Institute for
Branislav K. Nikolić
 First-principles quantum transport modeling of thermoelectricity in nanowires and single-molecule nanojunctions Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark,
First-principles quantum transport modeling of thermoelectricity in nanowires and single-molecule nanojunctions Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark,
A Review of Thermal Transport in Low- Dimensional Materials Under External Perturbation: Effect of Strain, Substrate, and Clustering
 Nanoscale and Microscale Thermophysical Engineering ISSN: 1556-7265 (Print) 1556-7273 (Online) Journal homepage: http://www.tandfonline.com/loi/umte20 A Review of Thermal Transport in Low- Dimensional
Nanoscale and Microscale Thermophysical Engineering ISSN: 1556-7265 (Print) 1556-7273 (Online) Journal homepage: http://www.tandfonline.com/loi/umte20 A Review of Thermal Transport in Low- Dimensional
Research Article Isotope Effect on the Thermal Conductivity of Graphene
 Nanomaterials Volume 2, Article ID 537657, 5 pages doi:.55/2/537657 Research Article Isotope Effect on the Thermal Conductivity of Graphene Hengji Zhang, Geunsik Lee, Alexandre F. Fonseca, 2 Tammie L.
Nanomaterials Volume 2, Article ID 537657, 5 pages doi:.55/2/537657 Research Article Isotope Effect on the Thermal Conductivity of Graphene Hengji Zhang, Geunsik Lee, Alexandre F. Fonseca, 2 Tammie L.
Complex superlattice unit cell designs for reduced thermal conductivity. Abstract
 Complex superlattice unit cell designs for reduced thermal conductivity E. S. Landry Department of Mechanical Engineering Carnegie Mellon University Pittsburgh, PA 15213 M. I. Hussein Department of Aerospace
Complex superlattice unit cell designs for reduced thermal conductivity E. S. Landry Department of Mechanical Engineering Carnegie Mellon University Pittsburgh, PA 15213 M. I. Hussein Department of Aerospace
arxiv: v1 [cond-mat.mes-hall] 29 Apr 2017
![arxiv: v1 [cond-mat.mes-hall] 29 Apr 2017](/thumbs/90/104470553.jpg "arxiv: v1 [cond-mat.mes-hall] 29 Apr 2017") Theoretical treatment of anharmonicity of vibrational modes of single-walled carbon nanotubes Hengjia Wang, 1 Doyl Dickel, 2 and Murray S. Daw 1 1 Department of Physics and Astronomy, arxiv:1705.00201v1
Theoretical treatment of anharmonicity of vibrational modes of single-walled carbon nanotubes Hengjia Wang, 1 Doyl Dickel, 2 and Murray S. Daw 1 1 Department of Physics and Astronomy, arxiv:1705.00201v1
Complex superlattice unit cell designs for reduced thermal conductivity
 Complex superlattice unit cell designs for reduced thermal conductivity E. S. Landry Department of Mechanical Engineering, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, USA M. I. Hussein
Complex superlattice unit cell designs for reduced thermal conductivity E. S. Landry Department of Mechanical Engineering, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, USA M. I. Hussein
PARALLEL MEASUREMENT OF CONDUCTIVE AND CONVECTIVE THERMAL TRANSPORT OF MICRO/NANOWIRES BASED ON RAMAN MAPPING
 Proceedings of the Asian Conference on Thermal Sciences 217, 1st ACTS March 26-3, 217, Jeju Island, Korea ACTS-4 PARALLEL MEASUREMENT OF CONDUCTIVE AND CONVECTIVE THERMAL TRANSPORT OF MICRO/NANOWIRES BASED
Proceedings of the Asian Conference on Thermal Sciences 217, 1st ACTS March 26-3, 217, Jeju Island, Korea ACTS-4 PARALLEL MEASUREMENT OF CONDUCTIVE AND CONVECTIVE THERMAL TRANSPORT OF MICRO/NANOWIRES BASED
Quantitatively Analyzing Phonon Spectral Contribution of Thermal. Conductivity Based on Non-Equilibrium Molecular Dynamics Simulation
 Quantitatively Analyzing Phonon Spectral Contribution of Thermal Conductivity Based on Non-Equilibrium Molecular Dynamics Simulation II: From Time Fourier Transform Yanguang Zhou 1 1, 2, * and Ming Hu
Quantitatively Analyzing Phonon Spectral Contribution of Thermal Conductivity Based on Non-Equilibrium Molecular Dynamics Simulation II: From Time Fourier Transform Yanguang Zhou 1 1, 2, * and Ming Hu
Edge effects on the electronic properties of phosphorene nanoribbons
 Edge effects on the electronic properties of phosphorene nanoribbons Xihong Peng, * Qun Wei,, Andrew Copple School of Letters and Sciences, Arizona State University, Mesa, Arizona 8, USA School of Physics
Edge effects on the electronic properties of phosphorene nanoribbons Xihong Peng, * Qun Wei,, Andrew Copple School of Letters and Sciences, Arizona State University, Mesa, Arizona 8, USA School of Physics
Enhancing thermal rectification in graphene-carbon nanotube junctions by tuning the chirality of pillar
 August 2018 EPL, 123 (2018) 44004 doi: 10.1209/0295-5075/123/44004 www.epljournal.org Enhancing thermal rectification in graphene-carbon nanotube junctions by tuning the chirality of pillar Xueming Yang
August 2018 EPL, 123 (2018) 44004 doi: 10.1209/0295-5075/123/44004 www.epljournal.org Enhancing thermal rectification in graphene-carbon nanotube junctions by tuning the chirality of pillar Xueming Yang
Effects of intrinsic strain on the structural stability and mechanical. properties of phosphorene nanotubes
 Effects of intrinsic strain on the structural stability and mechanical properties of phosphorene nanotubes Xiangbiao Liao 1, Feng Hao 1, Hang Xiao 1, and Xi Chen 1,2,* 1 Department of Earth and Environmental
Effects of intrinsic strain on the structural stability and mechanical properties of phosphorene nanotubes Xiangbiao Liao 1, Feng Hao 1, Hang Xiao 1, and Xi Chen 1,2,* 1 Department of Earth and Environmental
Effects of torsion on the thermal conductivity of multi-layer graphene
 JOURNAL OF APPLIED PHYSICS 121, 205102 (2017) Effects of torsion on the thermal conductivity of multi-layer graphene Chao Si, 1,2 Gui Lu, 1,2 Bing-Yang Cao, 3,a) Xiao-Dong Wang, 1,2,a) Zhen Fan, 4 and
JOURNAL OF APPLIED PHYSICS 121, 205102 (2017) Effects of torsion on the thermal conductivity of multi-layer graphene Chao Si, 1,2 Gui Lu, 1,2 Bing-Yang Cao, 3,a) Xiao-Dong Wang, 1,2,a) Zhen Fan, 4 and
Introduction to phonon transport
 Introduction to phonon transport Ivana Savić Tyndall National Institute, Cork, Ireland Materials for a Sustainable Energy Future Tutorials, Institute for Pure & Applied Mathematics, UCLA September 12,
Introduction to phonon transport Ivana Savić Tyndall National Institute, Cork, Ireland Materials for a Sustainable Energy Future Tutorials, Institute for Pure & Applied Mathematics, UCLA September 12,
A comprehensive analysis about thermal conductivity of multi-layer graphene with N-doping, -CH 3 group, and single vacancy
 JOURNAL OF APPLIED PHYSICS 123, 135101 (2018) A comprehensive analysis about thermal conductivity of multi-layer graphene with N-doping, -CH 3 group, and single vacancy Chao Si, 1,2,3 Liang Li, 1,2,3 Gui
JOURNAL OF APPLIED PHYSICS 123, 135101 (2018) A comprehensive analysis about thermal conductivity of multi-layer graphene with N-doping, -CH 3 group, and single vacancy Chao Si, 1,2,3 Liang Li, 1,2,3 Gui
Ferroelectricity and Antiferroelectricity in Elemental Group-V Monolayer Materials
 Ferroelectricity and Antiferroelectricity in Elemental Group-V Monolayer Materials Chengcheng Xiao 1, Fang Wang 1, Shengyuan A. Yang 2, Yunhao Lu 1 * 1 State Key Laboratory of Silicon Materials, School
Ferroelectricity and Antiferroelectricity in Elemental Group-V Monolayer Materials Chengcheng Xiao 1, Fang Wang 1, Shengyuan A. Yang 2, Yunhao Lu 1 * 1 State Key Laboratory of Silicon Materials, School
and strong interlayer quantum confinement
 Supporting Information GeP3: A small indirect band gap 2D crystal with high carrier mobility and strong interlayer quantum confinement Yu Jing 1,3, Yandong Ma 1, Yafei Li 2, *, Thomas Heine 1,3 * 1 Wilhelm-Ostwald-Institute
Supporting Information GeP3: A small indirect band gap 2D crystal with high carrier mobility and strong interlayer quantum confinement Yu Jing 1,3, Yandong Ma 1, Yafei Li 2, *, Thomas Heine 1,3 * 1 Wilhelm-Ostwald-Institute
High thermoelectric performance in two-dimensional graphyne sheets predicted by first-principles calculations
 High thermoelectric performance in two-dimensional graphyne sheets predicted by first-principles calculations Xiaojian Tan, Hezhu Shao, Tianqi Hu, Guoqiang Liu,* Jun Jiang and Haochuan Jiang The thermoelectric
High thermoelectric performance in two-dimensional graphyne sheets predicted by first-principles calculations Xiaojian Tan, Hezhu Shao, Tianqi Hu, Guoqiang Liu,* Jun Jiang and Haochuan Jiang The thermoelectric
3-month progress Report
 3-month progress Report Graphene Devices and Circuits Supervisor Dr. P.A Childs Table of Content Abstract... 1 1. Introduction... 1 1.1 Graphene gold rush... 1 1.2 Properties of graphene... 3 1.3 Semiconductor
3-month progress Report Graphene Devices and Circuits Supervisor Dr. P.A Childs Table of Content Abstract... 1 1. Introduction... 1 1.1 Graphene gold rush... 1 1.2 Properties of graphene... 3 1.3 Semiconductor
Phonon thermal properties of graphene from molecular dynamics using different potentials
 THE JOURNAL OF CHEMICAL PHYSICS 145, 134705 (2016) Phonon thermal properties of graphene from molecular dynamics using different potentials Ji-Hang Zou ( 邹济杭 ), Zhen-Qiang Ye ( 叶振强 ), and Bing-Yang Cao
THE JOURNAL OF CHEMICAL PHYSICS 145, 134705 (2016) Phonon thermal properties of graphene from molecular dynamics using different potentials Ji-Hang Zou ( 邹济杭 ), Zhen-Qiang Ye ( 叶振强 ), and Bing-Yang Cao
Supplementary Materials
 Supplementary Materials Atomistic Origin of Brittle Failure of Boron Carbide from Large Scale Reactive Dynamics Simulations; Suggestions toward Improved Ductility Qi An and William A. Goddard III * Materials
Supplementary Materials Atomistic Origin of Brittle Failure of Boron Carbide from Large Scale Reactive Dynamics Simulations; Suggestions toward Improved Ductility Qi An and William A. Goddard III * Materials
Electronic structure and properties of a few-layer black phosphorus Mikhail Katsnelson
 Electronic structure and properties of a few-layer black phosphorus Mikhail Katsnelson Main collaborators: Sasha Rudenko Shengjun Yuan Rafa Roldan Milton Pereira Sergey Brener Motivation Plenty of 2D materials
Electronic structure and properties of a few-layer black phosphorus Mikhail Katsnelson Main collaborators: Sasha Rudenko Shengjun Yuan Rafa Roldan Milton Pereira Sergey Brener Motivation Plenty of 2D materials
Shear Properties and Wrinkling Behaviors of Finite Sized Graphene
 Shear Properties and Wrinkling Behaviors of Finite Sized Graphene Kyoungmin Min, Namjung Kim and Ravi Bhadauria May 10, 2010 Abstract In this project, we investigate the shear properties of finite sized
Shear Properties and Wrinkling Behaviors of Finite Sized Graphene Kyoungmin Min, Namjung Kim and Ravi Bhadauria May 10, 2010 Abstract In this project, we investigate the shear properties of finite sized
Supporting Information for Solid-liquid Thermal Transport and its Relationship with Wettability and the Interfacial Liquid Structure
 Supporting Information for Solid-liquid Thermal Transport and its Relationship with Wettability and the Interfacial Liquid Structure Bladimir Ramos-Alvarado, Satish Kumar, and G. P. Peterson The George
Supporting Information for Solid-liquid Thermal Transport and its Relationship with Wettability and the Interfacial Liquid Structure Bladimir Ramos-Alvarado, Satish Kumar, and G. P. Peterson The George
arxiv: v1 [cond-mat.mes-hall] 28 Nov 2015
![arxiv: v1 [cond-mat.mes-hall] 28 Nov 2015](/thumbs/88/116630969.jpg "arxiv: v1 [cond-mat.mes-hall] 28 Nov 2015") Spectral energy analysis of locally resonant nanophononic metamaterials by molecular simulations Hossein Honarvar and Mahmoud I. Hussein Department of Aerospace Engineering Sciences, University of Colorado
Spectral energy analysis of locally resonant nanophononic metamaterials by molecular simulations Hossein Honarvar and Mahmoud I. Hussein Department of Aerospace Engineering Sciences, University of Colorado
Edinburgh Research Explorer
 Edinburgh Research Explorer Hydrogenation induced carrier mobility polarity reversal in single layer AlN Citation for published version: Ackland, G & Zong, H 2017, 'Hydrogenation induced carrier mobility
Edinburgh Research Explorer Hydrogenation induced carrier mobility polarity reversal in single layer AlN Citation for published version: Ackland, G & Zong, H 2017, 'Hydrogenation induced carrier mobility
Chapter 3 Properties of Nanostructures
 Chapter 3 Properties of Nanostructures In Chapter 2, the reduction of the extent of a solid in one or more dimensions was shown to lead to a dramatic alteration of the overall behavior of the solids. Generally,
Chapter 3 Properties of Nanostructures In Chapter 2, the reduction of the extent of a solid in one or more dimensions was shown to lead to a dramatic alteration of the overall behavior of the solids. Generally,
Transition of thermal rectification in silicon nanocones
 Transition of thermal rectification in silicon nanocones Zhongwei Zhang a, Yuanping Chen a*, Yuee Xie a, and Shengbai Zhang b a Department of Physics, Xiangtan University, Xiangtan 411105, Hunan, P.R.
Transition of thermal rectification in silicon nanocones Zhongwei Zhang a, Yuanping Chen a*, Yuee Xie a, and Shengbai Zhang b a Department of Physics, Xiangtan University, Xiangtan 411105, Hunan, P.R.
Misfit strain-induced energy dissipation for graphene/mos 2 heterostructure nanomechanical resonators. Abstract
 Misfit strain-induced energy dissipation for graphene/mos 2 heterostructure nanomechanical resonators Ji-Dong He and Jin-Wu Jiang arxiv:1901.03023v1 [cond-mat.mes-hall] 10 Jan 2019 Shanghai Institute of
Misfit strain-induced energy dissipation for graphene/mos 2 heterostructure nanomechanical resonators Ji-Dong He and Jin-Wu Jiang arxiv:1901.03023v1 [cond-mat.mes-hall] 10 Jan 2019 Shanghai Institute of
Ab initio molecular dynamics simulation on temperature-dependent properties of Al Si liquid alloy
 INSTITUTE OF PHYSICSPUBLISHING JOURNAL OFPHYSICS: CONDENSED MATTER J. Phys.: Condens. Matter 16 (4) 57 514 PII: S953-8984(4)7691-8 Ab initio molecular dynamics simulation on temperature-dependent properties
INSTITUTE OF PHYSICSPUBLISHING JOURNAL OFPHYSICS: CONDENSED MATTER J. Phys.: Condens. Matter 16 (4) 57 514 PII: S953-8984(4)7691-8 Ab initio molecular dynamics simulation on temperature-dependent properties
Minimum superlattice thermal conductivity from molecular dynamics
 Minimum superlattice thermal conductivity from molecular dynamics Yunfei Chen* Department of Mechanical Engineering and China Education Council Key Laboratory of MEMS, Southeast University, Nanjing, 210096,
Minimum superlattice thermal conductivity from molecular dynamics Yunfei Chen* Department of Mechanical Engineering and China Education Council Key Laboratory of MEMS, Southeast University, Nanjing, 210096,
Anisotropic Phonon Transport, Thermal Expansion and. Thermomechanics in Monolayer Td-WTe 2
 Anisotropic Phonon Transport, Thermal Expansion and Thermomechanics in Monolayer Td-WTe 2 Hongyi Sun 1, Qingfang Li 2* 1 National Laboratory of Solid State Microstructures, College of Physics, Nanjing
Anisotropic Phonon Transport, Thermal Expansion and Thermomechanics in Monolayer Td-WTe 2 Hongyi Sun 1, Qingfang Li 2* 1 National Laboratory of Solid State Microstructures, College of Physics, Nanjing
Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation
 Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation J. M. de Sousa 1,2, A. L. Aguiar 2, E. C. Girão 2, Alexandre F. Fonseca 1, A. G. Sousa Filho 3, and Douglas
Mechanical Properties of Phagraphene Membranes: A Fully Atomistic Molecular Dynamics Investigation J. M. de Sousa 1,2, A. L. Aguiar 2, E. C. Girão 2, Alexandre F. Fonseca 1, A. G. Sousa Filho 3, and Douglas
Multicomponent TMD Phase-field model with elastic heterogeneity
 Multicomponent TMD Phase-field model with elastic heterogeneity Yang Xia Department of Mechanical and Aerospace Engineering, Princeton University, Princeton, NJ 08544, USA Abstract A generalized semi 2D-model
Multicomponent TMD Phase-field model with elastic heterogeneity Yang Xia Department of Mechanical and Aerospace Engineering, Princeton University, Princeton, NJ 08544, USA Abstract A generalized semi 2D-model
Phonon thermal conduction in novel two-dimensional materials
 Phonon thermal conduction in novel two-dimensional materials Xiangfan Xu 1,2,3, Jie Chen 1,2,3, Baowen Li 4 1 Center for Phononics and Thermal Energy Science, School of Physics Science and Engineering,
Phonon thermal conduction in novel two-dimensional materials Xiangfan Xu 1,2,3, Jie Chen 1,2,3, Baowen Li 4 1 Center for Phononics and Thermal Energy Science, School of Physics Science and Engineering,
Recap (so far) Low-Dimensional & Boundary Effects
 Low-Dimensional & Boundary Effects") Recap (so far) Ohm s & Fourier s Laws Mobility & Thermal Conductivity Heat Capacity Wiedemann-Franz Relationship Size Effects and Breakdown of Classical Laws 1 Low-Dimensional & Boundary Effects Energy
Recap (so far) Ohm s & Fourier s Laws Mobility & Thermal Conductivity Heat Capacity Wiedemann-Franz Relationship Size Effects and Breakdown of Classical Laws 1 Low-Dimensional & Boundary Effects Energy
Nanosheet-Constructed Porous BiOCl with Dominant {001} Facets for Superior Photosensitized Degradation
 Electronic Supplementary Information Nanosheet-Constructed Porous BiOCl with Dominant {001} Facets for Superior Photosensitized Degradation Dong-Hong Wang, ab Gui-Qi Gao, b Yue-Wei Zhang, a Li-Sha Zhou,
Electronic Supplementary Information Nanosheet-Constructed Porous BiOCl with Dominant {001} Facets for Superior Photosensitized Degradation Dong-Hong Wang, ab Gui-Qi Gao, b Yue-Wei Zhang, a Li-Sha Zhou,
Session Chair: Prof. Haiping Cheng (University of Florida) Dr. Lei Shen. National University of Singapore
 Dr. Lei Shen. National University of Singapore") B1. Modeling Quantum Transport at Nanoscale Chair(s): Chun ZHANG, National University of Singapore, Singapore Session s Title (if available) Tue - 17 Jan 2017 13:00 ~ 14:30 Room 2 Session Chair: Prof.
B1. Modeling Quantum Transport at Nanoscale Chair(s): Chun ZHANG, National University of Singapore, Singapore Session s Title (if available) Tue - 17 Jan 2017 13:00 ~ 14:30 Room 2 Session Chair: Prof.
Supporting Information Tuning Local Electronic Structure of Single Layer MoS2 through Defect Engineering
 Supporting Information Tuning Local Electronic Structure of Single Layer MoS2 through Defect Engineering Yan Chen, 1,2,,$, * Shengxi Huang, 3,6, Xiang Ji, 2 Kiran Adepalli, 2 Kedi Yin, 8 Xi Ling, 3,9 Xinwei
Supporting Information Tuning Local Electronic Structure of Single Layer MoS2 through Defect Engineering Yan Chen, 1,2,,$, * Shengxi Huang, 3,6, Xiang Ji, 2 Kiran Adepalli, 2 Kedi Yin, 8 Xi Ling, 3,9 Xinwei
Two-dimensional Phosphorus Carbide as Promising Anode Materials for Lithium-ion Batteries
 Electronic Supplementary Material (ESI) for Journal of Materials Chemistry A. This journal is The Royal Society of Chemistry 2018 Supplementary Material for Two-dimensional Phosphorus Carbide as Promising
Electronic Supplementary Material (ESI) for Journal of Materials Chemistry A. This journal is The Royal Society of Chemistry 2018 Supplementary Material for Two-dimensional Phosphorus Carbide as Promising
Thermal characterization of Au-Si multilayer using 3- omega method
 Thermal characterization of Au-Si multilayer using 3- omega method Sunmi Shin Materials Science and Engineering Program Abstract As thermal management becomes a serious issue in applications of thermoelectrics,
Thermal characterization of Au-Si multilayer using 3- omega method Sunmi Shin Materials Science and Engineering Program Abstract As thermal management becomes a serious issue in applications of thermoelectrics,
arxiv: v1 [cond-mat.mes-hall] 15 Aug 2014
![arxiv: v1 [cond-mat.mes-hall] 15 Aug 2014](/thumbs/89/97831186.jpg "arxiv: v1 [cond-mat.mes-hall] 15 Aug 2014") The potential applications of phosphorene as anode arxiv:1408.3488v1 [cond-mat.mes-hall] 15 Aug 2014 materials in Li-ion batteries Shijun Zhao,, and Wei Kang, HEDPS, Center for Applied Physics and Technology,
The potential applications of phosphorene as anode arxiv:1408.3488v1 [cond-mat.mes-hall] 15 Aug 2014 materials in Li-ion batteries Shijun Zhao,, and Wei Kang, HEDPS, Center for Applied Physics and Technology,
Geometry Optimisation
 Geometry Optimisation Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, UK http://www.cmt.york.ac.uk/cmd http://www.castep.org Motivation Overview of Talk Background
Geometry Optimisation Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, UK http://www.cmt.york.ac.uk/cmd http://www.castep.org Motivation Overview of Talk Background
Quantum Effects and Phase Tuning in Epitaxial 2H- and 1T -MoTe 2 Monolayers
 Supplementary Information Quantum Effects and Phase Tuning in Epitaxial 2H- and 1T -MoTe 2 Monolayers Jinglei Chen, Guanyong Wang,, ǁ Yanan Tang,, Hao Tian,,# Jinpeng Xu, Xianqi Dai,, Hu Xu, # Jinfeng
Supplementary Information Quantum Effects and Phase Tuning in Epitaxial 2H- and 1T -MoTe 2 Monolayers Jinglei Chen, Guanyong Wang,, ǁ Yanan Tang,, Hao Tian,,# Jinpeng Xu, Xianqi Dai,, Hu Xu, # Jinfeng
First-Principles Prediction of Phononic Thermal Conductivity of Silicene: a. Comparison with Graphene
 First-Principles Prediction of Phononic Thermal Conductivity of Silicene: a Comparison with Graphene Xiaokun Gu and Ronggui Yang Department of Mechanical Engineering University of Colorado at Boulder,
First-Principles Prediction of Phononic Thermal Conductivity of Silicene: a Comparison with Graphene Xiaokun Gu and Ronggui Yang Department of Mechanical Engineering University of Colorado at Boulder,
(a) (b) Supplementary Figure 1. (a) (b) (a) Supplementary Figure 2. (a) (b) (c) (d) (e)
 (b) Supplementary Figure 1. (a) (b) (a) Supplementary Figure 2. (a) (b) (c) (d) (e)") (a) (b) Supplementary Figure 1. (a) An AFM image of the device after the formation of the contact electrodes and the top gate dielectric Al 2 O 3. (b) A line scan performed along the white dashed line
(a) (b) Supplementary Figure 1. (a) An AFM image of the device after the formation of the contact electrodes and the top gate dielectric Al 2 O 3. (b) A line scan performed along the white dashed line
An Extended Hückel Theory based Atomistic Model for Graphene Nanoelectronics
 Journal of Computational Electronics X: YYY-ZZZ,? 6 Springer Science Business Media, Inc. Manufactured in The Netherlands An Extended Hückel Theory based Atomistic Model for Graphene Nanoelectronics HASSAN
Journal of Computational Electronics X: YYY-ZZZ,? 6 Springer Science Business Media, Inc. Manufactured in The Netherlands An Extended Hückel Theory based Atomistic Model for Graphene Nanoelectronics HASSAN
PHONON TRANSPORT IN AMORPHOUS SILICON NANOWIRES. D.V. Crismari
 PHONON TRANSPORT IN AMORPHOUS SILICON NANOWIRES D.V. Crismari E. P. Pokatilov Laboratory of Physics and Engineering of Nanomaterials and Synergetics Moldova State University, Chisinau, Republic of Moldova
PHONON TRANSPORT IN AMORPHOUS SILICON NANOWIRES D.V. Crismari E. P. Pokatilov Laboratory of Physics and Engineering of Nanomaterials and Synergetics Moldova State University, Chisinau, Republic of Moldova
This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and
 This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution
This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution
Carbon based Nanoscale Electronics
 Carbon based Nanoscale Electronics 09 02 200802 2008 ME class Outline driving force for the carbon nanomaterial electronic properties of fullerene exploration of electronic carbon nanotube gold rush of
Carbon based Nanoscale Electronics 09 02 200802 2008 ME class Outline driving force for the carbon nanomaterial electronic properties of fullerene exploration of electronic carbon nanotube gold rush of
Strength and stability analysis of a single-walled black phosphorus tube under axial
 Home Search Collections Journals About Contact us My IOPscience Strength and stability analysis of a single-walled black phosphorus tube under axial compression This content has been downloaded from IOPscience.
Home Search Collections Journals About Contact us My IOPscience Strength and stability analysis of a single-walled black phosphorus tube under axial compression This content has been downloaded from IOPscience.
Nanoscale Energy Transport and Conversion A Parallel Treatment of Electrons, Molecules, Phonons, and Photons
 Nanoscale Energy Transport and Conversion A Parallel Treatment of Electrons, Molecules, Phonons, and Photons Gang Chen Massachusetts Institute of Technology OXFORD UNIVERSITY PRESS 2005 Contents Foreword,
Nanoscale Energy Transport and Conversion A Parallel Treatment of Electrons, Molecules, Phonons, and Photons Gang Chen Massachusetts Institute of Technology OXFORD UNIVERSITY PRESS 2005 Contents Foreword,
STRONG CONFIGURATIONAL DEPENDENCE OF ELASTIC PROPERTIES OF A CU-ZR BINARY MODEL METALLIC GLASS
 Chapter 3 STRONG CONFIGURATIONAL DEPENDENCE OF ELASTIC PROPERTIES OF A CU-ZR BINARY MODEL METALLIC GLASS We report the strong dependence of elastic properties on configurational changes in a Cu-Zr binary
Chapter 3 STRONG CONFIGURATIONAL DEPENDENCE OF ELASTIC PROPERTIES OF A CU-ZR BINARY MODEL METALLIC GLASS We report the strong dependence of elastic properties on configurational changes in a Cu-Zr binary
Carnegie Mellon University Carnegie Institute of Technology
 Carnegie Mellon University Carnegie Institute of Technology THESIS Submitted in Partial Fulfillment of the Requirements For the Degree of Doctor of Philosophy TITLE THERMAL TRANSPORT BY PHONONS ACROSS
Carnegie Mellon University Carnegie Institute of Technology THESIS Submitted in Partial Fulfillment of the Requirements For the Degree of Doctor of Philosophy TITLE THERMAL TRANSPORT BY PHONONS ACROSS
Nanoscale interfacial heat transfer: insights from molecular dynamics
 Nanoscale interfacial heat transfer: insights from molecular dynamics S. Merabia, A. Alkurdi, T. Albaret ILM CNRS and Université Lyon 1, France K.Termentzidis, D. Lacroix LEMTA, Université Lorraine, France
Nanoscale interfacial heat transfer: insights from molecular dynamics S. Merabia, A. Alkurdi, T. Albaret ILM CNRS and Université Lyon 1, France K.Termentzidis, D. Lacroix LEMTA, Université Lorraine, France
GeSi Quantum Dot Superlattices
 GeSi Quantum Dot Superlattices ECE440 Nanoelectronics Zheng Yang Department of Electrical & Computer Engineering University of Illinois at Chicago Nanostructures & Dimensionality Bulk Quantum Walls Quantum
GeSi Quantum Dot Superlattices ECE440 Nanoelectronics Zheng Yang Department of Electrical & Computer Engineering University of Illinois at Chicago Nanostructures & Dimensionality Bulk Quantum Walls Quantum
Department of Engineering Mechanics, SVL, Xi an Jiaotong University, Xi an
 The statistical characteristics of static friction J. Wang, G. F. Wang*, and W. K. Yuan Department of Engineering Mechanics, SVL, Xi an Jiaotong University, Xi an 710049, China * E-mail: wanggf@mail.xjtu.edu.cn
The statistical characteristics of static friction J. Wang, G. F. Wang*, and W. K. Yuan Department of Engineering Mechanics, SVL, Xi an Jiaotong University, Xi an 710049, China * E-mail: wanggf@mail.xjtu.edu.cn
Nanoscale PAPER. 1. Introduction. Jin-Wu Jiang,* a Timon Rabczuk b and Harold S. Park* c. View Article Online View Journal View Issue
 PAPER View Article Online View Journal View Issue Cite this:, 2015, 7, 6059 Received 12th December 2014, Accepted 26th February 2015 DOI: 10.1039/c4nr07341j www.rsc.org/nanoscale 1. Introduction A Stillinger
PAPER View Article Online View Journal View Issue Cite this:, 2015, 7, 6059 Received 12th December 2014, Accepted 26th February 2015 DOI: 10.1039/c4nr07341j www.rsc.org/nanoscale 1. Introduction A Stillinger
PCCP PAPER. Buckling behaviour of composites with double walled nanotubes from carbon and phosphorus. 1. Introduction. Kun Cai, ab Jing Wan,
 PAPER Cite this: Phys. Chem. Chem. Phys., 2017, 19, 10922 Buckling behaviour of composites with double walled nanotubes from carbon and phosphorus Kun Cai, ab Jing Wan, a Likui Yang, a Ning Wei,* a Jiao
PAPER Cite this: Phys. Chem. Chem. Phys., 2017, 19, 10922 Buckling behaviour of composites with double walled nanotubes from carbon and phosphorus Kun Cai, ab Jing Wan, a Likui Yang, a Ning Wei,* a Jiao
Supplemental material. The conflicting role of buckled structure in phonon. transport of 2D group-iv and group-v materials
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2017 Supplemental material The conflicting role of buckled structure in phonon transport of 2D group-iv
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2017 Supplemental material The conflicting role of buckled structure in phonon transport of 2D group-iv
Accuracy and transferability of GAP models for tungsten
 Accuracy and transferability of GAP models for tungsten Wojciech J. Szlachta Albert P. Bartók Gábor Csányi Engineering Laboratory University of Cambridge 5 November 214 Motivation Number of atoms 1 1 2
Accuracy and transferability of GAP models for tungsten Wojciech J. Szlachta Albert P. Bartók Gábor Csányi Engineering Laboratory University of Cambridge 5 November 214 Motivation Number of atoms 1 1 2
Evaluation of Electronic Characteristics of Double Gate Graphene Nanoribbon Field Effect Transistor for Wide Range of Temperatures
 Evaluation of Electronic Characteristics of Double Gate Graphene Nanoribbon Field Effect Transistor for Wide Range of Temperatures 1 Milad Abtin, 2 Ali Naderi 1 Department of electrical engineering, Masjed
Evaluation of Electronic Characteristics of Double Gate Graphene Nanoribbon Field Effect Transistor for Wide Range of Temperatures 1 Milad Abtin, 2 Ali Naderi 1 Department of electrical engineering, Masjed