Proteome-wide label-free quantification with MaxQuant. Jürgen Cox Max Planck Institute of Biochemistry July 2011
|
|
|
- Arnold Griffin
- 6 years ago
- Views:
Transcription
1 Proteome-wide label-free quantification with MaxQuant Jürgen Cox Max Planck Institute of Biochemistry July 2011


2 MaxQuant MaxQuant Feature detection Data acquisition Initial Andromeda search Statistics & systems biology Raw data Recalibration MQ output tables Main Andromeda search Consolidation / protein quantification Perseus Inspection of raw data Viewer
3 Supported input data Labeling methods Mass spectrometers SILAC Label free Di-methyl 18O ICAT ICPL Thermo Fisher Orbitrap and FT Work in progress: itraq Work in progress: SCIEX Triple TOF

4 Search engine MaxQuant Feature detection Initial Andromeda search Recalibration Peak lists Parameters Standalone Andromeda Single spectrum Peak list Andromeda web server Main Andromeda search Peptides Proteins Consolidation / protein quantification Visualization Cox et al, Andromeda a peptide search engine integrated into the MaxQuant environment. JPR (2011)

5 Mascot vs. Andromeda score Mascot score Andromeda score 95% 90% 75% 50% 0%
6 Identification of co-fragmented peptides

7 Absolute vs. relative quantification Absolute quantification: copy numbers for each protein Relative quantification: compare same protein in different sample


8 XIC vs. spectral count m/z 1


9 3D peak detection 2D peaks are assembled into 3D peaks Two 2D peaks in adjacent scans are connected when Δm < 7ppm Also next to nearest scan is checked
10 De-isotoping

11 Calculation of precise peptide masses Calculate precise mean and standard deviation for each peptide mass
![8 Nonlinear mass recalibration 6 Without lock mass Mass error [ppm] 4 2 0 With lock mass -2 300](/docs-images/78/78704699/images/12-0.jpg "500 700 900 1100 1300 m/z [Th] 0. 8 Relative frequency 0. 6 0. 4 0.")
12 8 Nonlinear mass recalibration 6 Without lock mass Mass error [ppm] With lock mass m/z [Th] 0. 8 Relative frequency Mass error [ppm]
13 Nonlinear mass recalibration 8 6 Mass error [ppm] Retention time [min]
14 Nonlinear mass recalibration 8 6 Mass error [ppm] Retention time [min]
![ΔM [ppm] Nonlinear mass recalibration 4 2 0 First Andromeda search with 20ppm mass tolerance and score threshold 80-2 300 500 m/z 700 [Th] 900 1100 ΔM [ppm] 4 2 0-2 300 500 m/z 700 [Th] 900 1100](/docs-images/78/78704699/images/15-0.jpg "Represent Δm as functions of m/z and t 4 2 0-2 2 0-2 76 78 80 82 84 86 88 90 t [min] Determine x positions for piecewise linear approximation. Initialize y values with 0.")
15 ΔM [ppm] Nonlinear mass recalibration First Andromeda search with 20ppm mass tolerance and score threshold m/z 700 [Th] ΔM [ppm] m/z 700 [Th] Represent Δm as functions of m/z and t t [min] Determine x positions for piecewise linear approximation. Initialize y values with t [min] 4 Minimize residual error 4 ΔM [ppm] m/z 700 [Th] t [min] Subtract recalibration functions from all measured peptides Perform the actual Andromeda search with small individualized mass tolerances
16 Mass error [ppm] Mass error [ppm] Mass error [ppm] Mass error [ppm] Nonlinear mass recalibration m/z [Th] m/z [Th] Retention time [min] Retention time [min]
17 Nonlinear mass recalibration a. 9 Mass error [ppm] b. 2 Mass error [ppm] Retention time [min] Retention time [min]

18 Improvement in mass accuracy
19 Problems in label free quantification Irreproducibility of retention time Incompatibility with pre-fractionation Quantification in a sample relies on MS/MS identification Identified peptides can be different in different samples



20 Two LC-MS runs Retention time alignment Peptides are matched by mass and retention time (only preliminary) Retention time difference between second and first LC-MS run Retention time in first LC-MS run Estimate of false positives from point densities in different regions

21 Retention time alignment Retention time difference between second and first LC-MS run Retention time in first LC-MS run



22 Matching between runs Identifcation transfer only between same or adjacend slices/fractions Transfering identifications after alignment increases base for quantitation by >100%
23 Label-free quantification: normalization Fraction A B C D E F : : : : Peptide P: I P,A (N) = N A,6 XIC A,6 + N A,7 XIC A,7 + N A,8 XIC A,8 I P,B (N) = N B,5 XIC B,5 + N B,6 XIC B,6 + N B,7 XIC B,7 + N B,8 XIC B,8 I P,C (N) = N C,7 XIC C,7 + N C,8 XIC C,8 + N C,9 XIC C,9 I P,D (N) = N D,5 XIC D,5 + N D,6 XIC D,6 + N D,7 XIC D,7 I P,E (N) = N E,6 XIC E,6 + N E,7 XIC E,7 I P,F (N) = N F,7 XIC F,7 + N F,8 XIC F,8 Peptide Q: I Q,A (N) = N A,14 XIC A,14 + N A,15 XIC A,15 + N A,16 XIC A,16 I Q,B (N) = N B,13 XIC B,13 + N B,14 XIC B,14 + N B,15 XIC B,15 + N B,16 XIC B,16 I Q,C (N) = N C,13 XIC C,13 + N C,14 XIC C,14 + N C,15 XIC C,15 I Q,D (N) = N D,14 XIC D,14 + N D,15 XIC D,15 I Q,E (N) = N E,14 XIC E,14 + N E,15 XIC E,15 + N E,16 XIC E,16 I Q,F (N) = N F,14 XIC F,14 + N F,15 XIC F,15 Peptide R: I R,A (N) = N A,21 XIC A,21 + N A,22 XIC A,22 I R,B (N) = N B,19 XIC B,19 + N B,20 XIC B,20 + N B,21 XIC B,21 I R,C (N) = N C,20 XIC C,20 + N C,21 XIC C,21 + N C,22 XIC C,22 I R,D (N) = N D,20 XIC D,20 + N D,21 XIC D,21 I R,E (N) = N E,19 XIC E,19 + N E,20 XIC E,20 + N E,21 XIC E,21 I R,F (N) = N F,20 XIC F,20 + N F,21 XIC F,21 I P,A 2 ( I P,B (N)) ( ) ( ) HP (N) = I log P,A (N) 2 I + log I P,C (N) + P,A (N) log I P,D (N) I ( Q,A (N) 2 ) log I Q,B (N) ( ) ( ) HQ (N) = I Q,A (N) 2 I + log I Q,C (N) + Q,A (N) log I Q,D (N) I ( R,A (N) 2 ) log I R,B (N) ( ) + ( ) HR (N) = I R,A (N) 2 I log I R,C (N) + R,A (N) log I R,D (N) H(N) = H P (N) + H Q (N) + H R (N) + other peptides 2 + other sample pairs 2 + other sample pairs 2 + other sample pairs
24 a. >P63208 MPSIKLQSSDGEIFEVDVEIAKQSVTIKTMLEDLGMKDEGDD DPVPLPNVNAAILKKVIQWCTHHKDDPPPPEDDENKEKRTDD IPVWDQEFLKVDQGTLFELILAANYLDIKGLLDVTCKTVANM IKGKTPEEIRKTFNIKNDFTEEEEAQVRKENQWCEEK Protein quantification d. A B r BA C r CA r CB b. Peptide P 1 P 2 P 3 P 4 P 5 P 6 P 7 Sequence LQSSDGEIFEVDVEIAK TMLEDLGMK VIQWCTHHK RTDDIPVWDQEFLK TVANMIK TPEEIRK NDFTEEEEAQVR D r DA r DB r DC E r EA r EB r EC r ED F r FA r FB r FC r FD r FE A B C D E F e. r BA = I B / I A r CA = I C / I A r CB = I C / I B r DA = I D / I A r DB = I D / I B r DC = I D / I C r EC = I E / I C r ED = I E / I D I F = 0 c. f. Sample P 1 P 2 P 3 P 4 P 5 P 6 P 7 A + + B C D E F + + Intensity 0 A B C D E F
25 Label-free quantification Benchmark dataset HeLa and E. coli cell lysates are mixed Proteins were digested with trypsin. In three replicates peptides were separated by isoelectric focusing in 24 fractions. This was repeated with the same amount of HeLa, but E. coli lysate tripled. This results in six samples for which all human proteins have constant protein profiles, while E. coli proteins have a ratio of three between replicate groups. LC-MS on an LTQ-Orbitrap mass spectrometer. Data: Christian Luber
26 Identification results 1,234,125 MS isotope patterns identified by MS/MS 1,852,556 MS isotope patterns identified by matching between runs 3,086,681 MS isotope patterns in total 6,577 proteins 5,161 proteins in at least 3/6 samples 4,589 proteins in 6/6 samples 46,839 peptide sequences
")
27 Label-free quantification results Log(intensity) Log(ratio)
28 Dynamic range benchmark dataset

29 Comparison to SILAC 10 Protein ratio e5 1e6 1e7 1e8 1e9 1e10 1e11 Summed peptide intensity
30 Precision vs. recall Precision = TP / (TP + FP) t-test Welch modified t-test Wilcoxon-Mann-Whitney test ratio Recall = TP / (TP + FN)
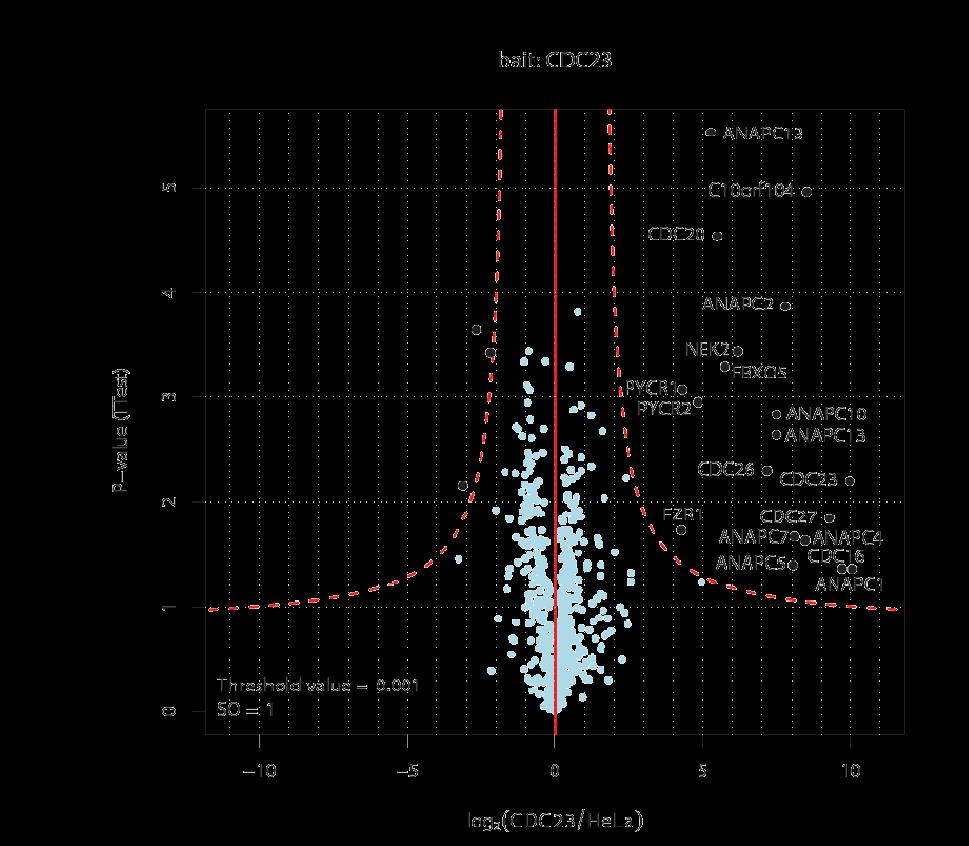
31 Pulldowns
32 Imputation Log(intensity)
33
34 groups.google.com/group/maxquant-list

35 Usability, documentation, software quality

36 Acknowledgements Matthias Mann Nadin Neuhauser Richard Scheltema Christoph Schaab Christian Luber All Mann lab members Thank you for your attention
Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ* S
 Author s Choice Technological Innovation and Resources 2014 by The American Society for Biochemistry and Molecular Biology, Inc. This paper is available on line at http://www.mcponline.org Accurate Proteome-wide
Author s Choice Technological Innovation and Resources 2014 by The American Society for Biochemistry and Molecular Biology, Inc. This paper is available on line at http://www.mcponline.org Accurate Proteome-wide
Workflow concept. Data goes through the workflow. A Node contains an operation An edge represents data flow The results are brought together in tables
 PROTEOME DISCOVERER Workflow concept Data goes through the workflow Spectra Peptides Quantitation A Node contains an operation An edge represents data flow The results are brought together in tables Protein
PROTEOME DISCOVERER Workflow concept Data goes through the workflow Spectra Peptides Quantitation A Node contains an operation An edge represents data flow The results are brought together in tables Protein
Mass spectrometry-based proteomics has become
 FOCUS: THE ORBITRAP Computational Principles of Determining and Improving Mass Precision and Accuracy for Proteome Measurements in an Orbitrap Jürgen Cox and Matthias Mann Proteomics and Signal Transduction,
FOCUS: THE ORBITRAP Computational Principles of Determining and Improving Mass Precision and Accuracy for Proteome Measurements in an Orbitrap Jürgen Cox and Matthias Mann Proteomics and Signal Transduction,
Overview - MS Proteomics in One Slide. MS masses of peptides. MS/MS fragments of a peptide. Results! Match to sequence database
 Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
Computational Methods for Mass Spectrometry Proteomics
 Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
Workshop: SILAC and Alternative Labeling Strategies in Quantitative Proteomics
 Workshop: SILAC and Alternative Labeling Strategies in Quantitative Proteomics SILAC and Stable Isotope Dimethyl-Labeling Approaches in Quantitative Proteomics Ho-Tak Lau, Hyong-Won Suh, Shao-En Ong UW
Workshop: SILAC and Alternative Labeling Strategies in Quantitative Proteomics SILAC and Stable Isotope Dimethyl-Labeling Approaches in Quantitative Proteomics Ho-Tak Lau, Hyong-Won Suh, Shao-En Ong UW
Comprehensive support for quantitation
 Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
Designed for Accuracy. Innovation with Integrity. High resolution quantitative proteomics LC-MS
 Designed for Accuracy High resolution quantitative proteomics Innovation with Integrity LC-MS Setting New Standards in Accuracy The development of mass spectrometry based proteomics approaches has dramatically
Designed for Accuracy High resolution quantitative proteomics Innovation with Integrity LC-MS Setting New Standards in Accuracy The development of mass spectrometry based proteomics approaches has dramatically
Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University
 Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Proteomics Sample prep 144 Lecture 5 Quantitation techniques Search Algorithms Proteomics
Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Proteomics Sample prep 144 Lecture 5 Quantitation techniques Search Algorithms Proteomics
Modeling Mass Spectrometry-Based Protein Analysis
 Chapter 8 Jan Eriksson and David Fenyö Abstract The success of mass spectrometry based proteomics depends on efficient methods for data analysis. These methods require a detailed understanding of the information
Chapter 8 Jan Eriksson and David Fenyö Abstract The success of mass spectrometry based proteomics depends on efficient methods for data analysis. These methods require a detailed understanding of the information
Analysis of Labeled and Non-Labeled Proteomic Data Using Progenesis QI for Proteomics
 Analysis of Labeled and Non-Labeled Proteomic Data Using Progenesis QI for Proteomics Lee Gethings, Gushinder Atwal, Martin Palmer, Chris Hughes, Hans Vissers, and James Langridge Waters Corporation, Wilmslow,
Analysis of Labeled and Non-Labeled Proteomic Data Using Progenesis QI for Proteomics Lee Gethings, Gushinder Atwal, Martin Palmer, Chris Hughes, Hans Vissers, and James Langridge Waters Corporation, Wilmslow,
Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 *
 Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 * 1 Department of Chemistry, Pomona College, Claremont, California
Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 * 1 Department of Chemistry, Pomona College, Claremont, California
Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics
 Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics Xiao-jun Li, Ph.D. Current address: Homestead Clinical Day 4 October 19, 2006 Protein Quantification LC-MS/MS Data XLink mzxml file
Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics Xiao-jun Li, Ph.D. Current address: Homestead Clinical Day 4 October 19, 2006 Protein Quantification LC-MS/MS Data XLink mzxml file
HR/AM Targeted Peptide Quantification on a Q Exactive MS: A Unique Combination of High Selectivity, High Sensitivity, and High Throughput
 HR/AM Targeted Peptide Quantification on a Q Exactive MS: A Unique Combination of High Selectivity, High Sensitivity, and High Throughput Yi Zhang 1, Zhiqi Hao 1, Markus Kellmann 2 and Andreas FR. Huhmer
HR/AM Targeted Peptide Quantification on a Q Exactive MS: A Unique Combination of High Selectivity, High Sensitivity, and High Throughput Yi Zhang 1, Zhiqi Hao 1, Markus Kellmann 2 and Andreas FR. Huhmer
MS-based proteomics to investigate proteins and their modifications
 MS-based proteomics to investigate proteins and their modifications Francis Impens VIB Proteomics Core October th 217 Overview Mass spectrometry-based proteomics: general workflow Identification of protein
MS-based proteomics to investigate proteins and their modifications Francis Impens VIB Proteomics Core October th 217 Overview Mass spectrometry-based proteomics: general workflow Identification of protein
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
Increasing the Multiplexing of Protein Quantitation from 6- to 10-Plex with Reporter Ion Isotopologues
 Increasing the Multiplexing of Protein Quantitation from 6- to 1-Plex with Reporter Ion Isotopologues Rosa Viner, 1 Ryan Bomgarden, 2 Michael Blank, 1 John Rogers 2 1 Thermo Fisher Scientific, San Jose,
Increasing the Multiplexing of Protein Quantitation from 6- to 1-Plex with Reporter Ion Isotopologues Rosa Viner, 1 Ryan Bomgarden, 2 Michael Blank, 1 John Rogers 2 1 Thermo Fisher Scientific, San Jose,
Relative quantification using TMT11plex on a modified Q Exactive HF mass spectrometer
 POSTER NOTE 6558 Relative quantification using TMT11plex on a modified mass spectrometer Authors Tabiwang N. Arrey, 1 Rosa Viner, 2 Ryan D. Bomgarden, 3 Eugen Damoc, 1 Markus Kellmann, 1 Thomas Moehring,
POSTER NOTE 6558 Relative quantification using TMT11plex on a modified mass spectrometer Authors Tabiwang N. Arrey, 1 Rosa Viner, 2 Ryan D. Bomgarden, 3 Eugen Damoc, 1 Markus Kellmann, 1 Thomas Moehring,
Spectronaut Pulsar. User Manual
 Spectronaut Pulsar User Manual 1 General Information... 6 1.1 Computer System Requirements... 6 1.2 Scope of Spectronaut Software... 6 1.3 Spectronaut Pulsar... 6 1.4 Spectronaut Release Features... 7
Spectronaut Pulsar User Manual 1 General Information... 6 1.1 Computer System Requirements... 6 1.2 Scope of Spectronaut Software... 6 1.3 Spectronaut Pulsar... 6 1.4 Spectronaut Release Features... 7
Relative Quantitation of TMT-Labeled Proteomes Focus on Sensitivity and Precision
 Relative Quantitation of TMT-Labeled Proteomes Focus on Sensitivity and Precision R. Viner 1, M. Scigelova 2, M. Zeller 2, M. Oppermann 2, T. Moehring 2 and V. Zabrouskov 1 1 Thermo Fisher Scientific,
Relative Quantitation of TMT-Labeled Proteomes Focus on Sensitivity and Precision R. Viner 1, M. Scigelova 2, M. Zeller 2, M. Oppermann 2, T. Moehring 2 and V. Zabrouskov 1 1 Thermo Fisher Scientific,
6 x 5 Ways to Ensure Your LC-MS/MS is Healthy
 6 x 5 Ways to Ensure Your LC-MS/MS is Healthy (Also known as - Tracking Performance with the 6 x 5 LC-MS/MS Peptide Reference Mixture) Mike Rosenblatt, Ph.D. Group Leader Mass Spec Reagents 215. We monitor
6 x 5 Ways to Ensure Your LC-MS/MS is Healthy (Also known as - Tracking Performance with the 6 x 5 LC-MS/MS Peptide Reference Mixture) Mike Rosenblatt, Ph.D. Group Leader Mass Spec Reagents 215. We monitor
NEW TOOLS FOR FINDING AND IDENTIFYING METABOLITES IN A METABOLOMICS WORKFLOW
 NEW TOOLS FOR FINDING AND IDENTIFYING METABOLITES IN A METABOLOMICS WORKFLOW Julia E. Wingate 1 ; Elliott Jones 2 ; Armin Graber 3 ; Klaus Weinberger 3 1Applied Biosystems, Toronto, Canada; 2Applied Biosystems,
NEW TOOLS FOR FINDING AND IDENTIFYING METABOLITES IN A METABOLOMICS WORKFLOW Julia E. Wingate 1 ; Elliott Jones 2 ; Armin Graber 3 ; Klaus Weinberger 3 1Applied Biosystems, Toronto, Canada; 2Applied Biosystems,
PC235: 2008 Lecture 5: Quantitation. Arnold Falick
 PC235: 2008 Lecture 5: Quantitation Arnold Falick falickam@berkeley.edu Summary What you will learn from this lecture: There are many methods to perform quantitation using mass spectrometry (any method
PC235: 2008 Lecture 5: Quantitation Arnold Falick falickam@berkeley.edu Summary What you will learn from this lecture: There are many methods to perform quantitation using mass spectrometry (any method
Methods for proteome analysis of obesity (Adipose tissue)
") Methods for proteome analysis of obesity (Adipose tissue) I. Sample preparation and liquid chromatography-tandem mass spectrometric analysis Instruments, softwares, and materials AB SCIEX Triple TOF 5600
Methods for proteome analysis of obesity (Adipose tissue) I. Sample preparation and liquid chromatography-tandem mass spectrometric analysis Instruments, softwares, and materials AB SCIEX Triple TOF 5600
Overview. Introduction. André Schreiber 1 and Yun Yun Zou 1 1 AB SCIEX, Concord, Ontario, Canada
 LC-MS/MS Based Strategy for the Non-Targeted Screening of an Unlimited Number of Contaminants in Food Using the AB SCIEX TripleTOF 5600 System and Advanced Software Tools André Schreiber 1 and Yun Yun
LC-MS/MS Based Strategy for the Non-Targeted Screening of an Unlimited Number of Contaminants in Food Using the AB SCIEX TripleTOF 5600 System and Advanced Software Tools André Schreiber 1 and Yun Yun
A Quadrupole-Orbitrap Hybrid Mass Spectrometer Offers Highest Benchtop Performance for In-Depth Analysis of Complex Proteomes
 A Quadrupole-Orbitrap Hybrid Mass Spectrometer Offers Highest Benchtop Performance for In-Depth Analysis of Complex Proteomes Zhiqi Hao 1, Yi Zhang 1, Shannon Eliuk 1, Justin Blethrow 1, Dave Horn 1, Vlad
A Quadrupole-Orbitrap Hybrid Mass Spectrometer Offers Highest Benchtop Performance for In-Depth Analysis of Complex Proteomes Zhiqi Hao 1, Yi Zhang 1, Shannon Eliuk 1, Justin Blethrow 1, Dave Horn 1, Vlad
UCD Conway Institute of Biomolecular & Biomedical Research Graduate Education 2009/2010
 EMERGING PROTEOMIC TECHNOLOGIES - MODULE SCHEDULE & OUTLINE 2010 Course Organiser: Dr. Giuliano Elia Module Co-ordinator: Dr Giuliano Elia Credits: 5 Date & Time Session & Topic Coordinator 14th April
EMERGING PROTEOMIC TECHNOLOGIES - MODULE SCHEDULE & OUTLINE 2010 Course Organiser: Dr. Giuliano Elia Module Co-ordinator: Dr Giuliano Elia Credits: 5 Date & Time Session & Topic Coordinator 14th April
Quantitative Proteomics
 BSPR workshop 16 th July 2010 Quantitative Proteomics Kathryn Lilley Cambridge Centre for Proteomics Department of Biochemistry University of Cambridge k.s.lilley@bioc.cam.ac.uk www.bio.cam.ac.uk/proteomics/
BSPR workshop 16 th July 2010 Quantitative Proteomics Kathryn Lilley Cambridge Centre for Proteomics Department of Biochemistry University of Cambridge k.s.lilley@bioc.cam.ac.uk www.bio.cam.ac.uk/proteomics/
DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics
 DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics Chih-Chiang Tsou 1,2, Dmitry Avtonomov 2, Brett Larsen 3, Monika Tucholska 3, Hyungwon Choi 4 Anne-Claude Gingras
DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics Chih-Chiang Tsou 1,2, Dmitry Avtonomov 2, Brett Larsen 3, Monika Tucholska 3, Hyungwon Choi 4 Anne-Claude Gingras
Targeted Proteomics Environment
 Targeted Proteomics Environment Quantitative Proteomics with Bruker Q-TOF Instruments and Skyline Brendan MacLean Quantitative Proteomics Spectrum-based Spectral counting Isobaric tags Chromatography-based
Targeted Proteomics Environment Quantitative Proteomics with Bruker Q-TOF Instruments and Skyline Brendan MacLean Quantitative Proteomics Spectrum-based Spectral counting Isobaric tags Chromatography-based
Chemical Labeling Strategy for Generation of Internal Standards for Targeted Quantitative Proteomics
 Chemical Labeling Strategy for Generation of Internal Standards for Targeted Quantitative Proteomics mtraq Reagents Triplex Christie Hunter, Brian Williamson, Marjorie Minkoff AB SCIEX, USA The utility
Chemical Labeling Strategy for Generation of Internal Standards for Targeted Quantitative Proteomics mtraq Reagents Triplex Christie Hunter, Brian Williamson, Marjorie Minkoff AB SCIEX, USA The utility
HOWTO, example workflow and data files. (Version )
") HOWTO, example workflow and data files. (Version 20 09 2017) 1 Introduction: SugarQb is a collection of software tools (Nodes) which enable the automated identification of intact glycopeptides from HCD
HOWTO, example workflow and data files. (Version 20 09 2017) 1 Introduction: SugarQb is a collection of software tools (Nodes) which enable the automated identification of intact glycopeptides from HCD
High-Field Orbitrap Creating new possibilities
 Thermo Scientific Orbitrap Elite Hybrid Mass Spectrometer High-Field Orbitrap Creating new possibilities Ultrahigh resolution Faster scanning Higher sensitivity Complementary fragmentation The highest
Thermo Scientific Orbitrap Elite Hybrid Mass Spectrometer High-Field Orbitrap Creating new possibilities Ultrahigh resolution Faster scanning Higher sensitivity Complementary fragmentation The highest
Thermo Scientific LTQ Orbitrap Velos Hybrid FT Mass Spectrometer
 IET International Equipment Trading Ltd. www.ietltd.com Proudly serving laboratories worldwide since 1979 CALL +847.913.0777 for Refurbished & Certified Lab Equipment Thermo Scientific LTQ Orbitrap Velos
IET International Equipment Trading Ltd. www.ietltd.com Proudly serving laboratories worldwide since 1979 CALL +847.913.0777 for Refurbished & Certified Lab Equipment Thermo Scientific LTQ Orbitrap Velos
NPTEL VIDEO COURSE PROTEOMICS PROF. SANJEEVA SRIVASTAVA
 LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
profileanalysis Innovation with Integrity Quickly pinpointing and identifying potential biomarkers in Proteomics and Metabolomics research
 profileanalysis Quickly pinpointing and identifying potential biomarkers in Proteomics and Metabolomics research Innovation with Integrity Omics Research Biomarker Discovery Made Easy by ProfileAnalysis
profileanalysis Quickly pinpointing and identifying potential biomarkers in Proteomics and Metabolomics research Innovation with Integrity Omics Research Biomarker Discovery Made Easy by ProfileAnalysis
Tutorial 2: Analysis of DIA data in Skyline
 Tutorial 2: Analysis of DIA data in Skyline In this tutorial we will learn how to use Skyline to perform targeted post-acquisition analysis for peptide and inferred protein detection and quantitation using
Tutorial 2: Analysis of DIA data in Skyline In this tutorial we will learn how to use Skyline to perform targeted post-acquisition analysis for peptide and inferred protein detection and quantitation using
Rapid Distinction of Leucine and Isoleucine in Monoclonal Antibodies Using Nanoflow. LCMS n. Discovery Attribute Sciences
 Rapid Distinction of Leucine and Isoleucine in Monoclonal Antibodies Using Nanoflow LCMS n Dhanashri Bagal *, Eddie Kast, Ping Cao Discovery Attribute Sciences Amgen, South San Francisco, California, United
Rapid Distinction of Leucine and Isoleucine in Monoclonal Antibodies Using Nanoflow LCMS n Dhanashri Bagal *, Eddie Kast, Ping Cao Discovery Attribute Sciences Amgen, South San Francisco, California, United
Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent
 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent") Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent concentrations of PcTS (100 µm, blue; 500 µm, green; 1.5 mm,
Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent concentrations of PcTS (100 µm, blue; 500 µm, green; 1.5 mm,
Statistical mass spectrometry-based proteomics
 1 Statistical mass spectrometry-based proteomics Olga Vitek www.stat.purdue.edu Outline What is proteomics? Biological questions and technologies Protein quantification in label-free workflows Joint analysis
1 Statistical mass spectrometry-based proteomics Olga Vitek www.stat.purdue.edu Outline What is proteomics? Biological questions and technologies Protein quantification in label-free workflows Joint analysis
Quantitative Proteomics
 Quantitative Proteomics Quantitation AND Mass Spectrometry Condition A Condition B Identify and quantify differently expressed proteins resulting from a change in the environment (stimulus, disease) Lyse
Quantitative Proteomics Quantitation AND Mass Spectrometry Condition A Condition B Identify and quantify differently expressed proteins resulting from a change in the environment (stimulus, disease) Lyse
De novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra. Xiaowen Liu
 De novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra Xiaowen Liu Department of BioHealth Informatics, Department of Computer and Information Sciences, Indiana University-Purdue
De novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra Xiaowen Liu Department of BioHealth Informatics, Department of Computer and Information Sciences, Indiana University-Purdue
Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment
 pubs.acs.org/jpr Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment J urgen Cox,*, Nadin Neuhauser, Annette Michalski, Richard A. Scheltema, Jesper V. Olsen, and Matthias Mann*,,
pubs.acs.org/jpr Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment J urgen Cox,*, Nadin Neuhauser, Annette Michalski, Richard A. Scheltema, Jesper V. Olsen, and Matthias Mann*,,
Protein analysis using mass spectrometry
 Protein analysis using mass spectrometry Michael Stadlmeier 2017/12/18 Literature http://www.carellgroup.de/teaching/master 3 What is Proteomics? The proteome is: the entire set of proteins in a given
Protein analysis using mass spectrometry Michael Stadlmeier 2017/12/18 Literature http://www.carellgroup.de/teaching/master 3 What is Proteomics? The proteome is: the entire set of proteins in a given
Self-assembling covalent organic frameworks functionalized. magnetic graphene hydrophilic biocomposite as an ultrasensitive
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2017 Electronic Supporting Information for: Self-assembling covalent organic frameworks functionalized
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2017 Electronic Supporting Information for: Self-assembling covalent organic frameworks functionalized
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry
 Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
Key questions of proteomics. Bioinformatics 2. Proteomics. Foundation of proteomics. What proteins are there? Protein digestion
 s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
Proteomics: the first decade and beyond. (2003) Patterson and Aebersold Nat Genet 33 Suppl: from
 Patterson and Aebersold Nat Genet 33 Suppl: from") Advances in mass spectrometry and the generation of large quantities of nucleotide sequence information, combined with computational algorithms that could correlate the two, led to the emergence of proteomics
Advances in mass spectrometry and the generation of large quantities of nucleotide sequence information, combined with computational algorithms that could correlate the two, led to the emergence of proteomics
Peptide Targeted Quantification By High Resolution Mass Spectrometry A Paradigm Shift? Zhiqi Hao Thermo Fisher Scientific San Jose, CA
 Peptide Targeted Quantification By High Resolution Mass Spectrometry A Paradigm Shift? Zhiqi Hao Thermo Fisher Scientific San Jose, CA Proteomics is Turning Quantitative Hmmm.. Which ones are my targets?
Peptide Targeted Quantification By High Resolution Mass Spectrometry A Paradigm Shift? Zhiqi Hao Thermo Fisher Scientific San Jose, CA Proteomics is Turning Quantitative Hmmm.. Which ones are my targets?
SeqAn and OpenMS Integration Workshop. Temesgen Dadi, Julianus Pfeuffer, Alexander Fillbrunn The Center for Integrative Bioinformatics (CIBI)
") SeqAn and OpenMS Integration Workshop Temesgen Dadi, Julianus Pfeuffer, Alexander Fillbrunn The Center for Integrative Bioinformatics (CIBI) Mass-spectrometry data analysis in KNIME Julianus Pfeuffer,
SeqAn and OpenMS Integration Workshop Temesgen Dadi, Julianus Pfeuffer, Alexander Fillbrunn The Center for Integrative Bioinformatics (CIBI) Mass-spectrometry data analysis in KNIME Julianus Pfeuffer,
TUTORIAL EXERCISES WITH ANSWERS
 TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
Atomic masses. Atomic masses of elements. Atomic masses of isotopes. Nominal and exact atomic masses. Example: CO, N 2 ja C 2 H 4
 High-Resolution Mass spectrometry (HR-MS, HRAM-MS) (FT mass spectrometry) MS that enables identifying elemental compositions (empirical formulas) from accurate m/z data 9.05.2017 1 Atomic masses (atomic
High-Resolution Mass spectrometry (HR-MS, HRAM-MS) (FT mass spectrometry) MS that enables identifying elemental compositions (empirical formulas) from accurate m/z data 9.05.2017 1 Atomic masses (atomic
Q Exactive TM : A True Qual-Quan HR/AM Mass Spectrometer for Routine Proteomics Applications. Yi Zhang, Ph.D. ThermoFisher Scientific
 Q Exactive TM : A True Qual-Quan HR/AM Mass Spectrometer for Routine Proteomics Applications Yi Zhang, Ph.D. ThermoFisher Scientific Outline Introduction of Q Exactive Performance in Discovery Proteomics
Q Exactive TM : A True Qual-Quan HR/AM Mass Spectrometer for Routine Proteomics Applications Yi Zhang, Ph.D. ThermoFisher Scientific Outline Introduction of Q Exactive Performance in Discovery Proteomics
Site-specific Identification of Lysine Acetylation Stoichiometries in Mammalian Cells
 Supplementary Information Site-specific Identification of Lysine Acetylation Stoichiometries in Mammalian Cells Tong Zhou 1, 2, Ying-hua Chung 1, 2, Jianji Chen 1, Yue Chen 1 1. Department of Biochemistry,
Supplementary Information Site-specific Identification of Lysine Acetylation Stoichiometries in Mammalian Cells Tong Zhou 1, 2, Ying-hua Chung 1, 2, Jianji Chen 1, Yue Chen 1 1. Department of Biochemistry,
Key Words Q Exactive, Accela, MetQuest, Mass Frontier, Drug Discovery
 Metabolite Stability Screening and Hotspot Metabolite Identification by Combining High-Resolution, Accurate-Mass Nonselective and Selective Fragmentation Tim Stratton, Caroline Ding, Yingying Huang, Dan
Metabolite Stability Screening and Hotspot Metabolite Identification by Combining High-Resolution, Accurate-Mass Nonselective and Selective Fragmentation Tim Stratton, Caroline Ding, Yingying Huang, Dan
pparse: A method for accurate determination of monoisotopic peaks in high-resolution mass spectra
 226 DOI 10.1002/pmic.201100081 Proteomics 2012, 12, 226 235 RESEARCH ARTICLE pparse: A method for accurate determination of monoisotopic peaks in high-resolution mass spectra Zuo-Fei Yuan 1,2, Chao Liu
226 DOI 10.1002/pmic.201100081 Proteomics 2012, 12, 226 235 RESEARCH ARTICLE pparse: A method for accurate determination of monoisotopic peaks in high-resolution mass spectra Zuo-Fei Yuan 1,2, Chao Liu
Quantitation of TMT-Labeled Peptides Using Higher-Energy Collisional Dissociation on the Velos Pro Ion Trap Mass Spectrometer
 Application Note: 520 Quantitation of TMT-Labeled Peptides Using Higher-Energy Collisional Dissociation on the Velos Pro Ion Trap Mass Spectrometer Roger G. Biringer, Julie A. Horner, Rosa Viner, Andreas
Application Note: 520 Quantitation of TMT-Labeled Peptides Using Higher-Energy Collisional Dissociation on the Velos Pro Ion Trap Mass Spectrometer Roger G. Biringer, Julie A. Horner, Rosa Viner, Andreas
Improved Throughput and Reproducibility for Targeted Protein Quantification Using a New High-Performance Triple Quadrupole Mass Spectrometer
 Improved Throughput and Reproducibility for Targeted Protein Quantification Using a New High-Performance Triple Quadrupole Mass Spectrometer Reiko Kiyonami, Mary Blackburn, Andreas FR Hühme: Thermo Fisher
Improved Throughput and Reproducibility for Targeted Protein Quantification Using a New High-Performance Triple Quadrupole Mass Spectrometer Reiko Kiyonami, Mary Blackburn, Andreas FR Hühme: Thermo Fisher
MassHunter TOF/QTOF Users Meeting
 MassHunter TOF/QTOF Users Meeting 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks
MassHunter TOF/QTOF Users Meeting 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks
iprophet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates
 MCP Papers in Press. Published on August 29, 2011 as Manuscript M111.007690 This is the Pre-Published Version iprophet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein
MCP Papers in Press. Published on August 29, 2011 as Manuscript M111.007690 This is the Pre-Published Version iprophet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein
Multi-residue analysis of pesticides by GC-HRMS
 An Executive Summary Multi-residue analysis of pesticides by GC-HRMS Dr. Hans Mol is senior scientist at RIKILT- Wageningen UR Introduction Regulatory authorities throughout the world set and enforce strict
An Executive Summary Multi-residue analysis of pesticides by GC-HRMS Dr. Hans Mol is senior scientist at RIKILT- Wageningen UR Introduction Regulatory authorities throughout the world set and enforce strict
MS Based Proteomics: Recent Case Studies Using Advanced Instrumentation
 MS Based Proteomics: Recent Case Studies Using Advanced Instrumentation Chris Adams, PH.D. Stanford University Mass Spectrometry http://mass-spec.stanford.edu/ For personal use only. Please do not reuse
MS Based Proteomics: Recent Case Studies Using Advanced Instrumentation Chris Adams, PH.D. Stanford University Mass Spectrometry http://mass-spec.stanford.edu/ For personal use only. Please do not reuse
A Description of the CPTAC Common Data Analysis Pipeline (CDAP)
") A Description of the CPTAC Common Data Analysis Pipeline (CDAP) v. 01/14/2014 Summary The purpose of this document is to describe the software programs and output files of the Common Data Analysis Pipeline
A Description of the CPTAC Common Data Analysis Pipeline (CDAP) v. 01/14/2014 Summary The purpose of this document is to describe the software programs and output files of the Common Data Analysis Pipeline
Improved Validation of Peptide MS/MS Assignments. Using Spectral Intensity Prediction
 MCP Papers in Press. Published on October 2, 2006 as Manuscript M600320-MCP200 Improved Validation of Peptide MS/MS Assignments Using Spectral Intensity Prediction Shaojun Sun 1, Karen Meyer-Arendt 2,
MCP Papers in Press. Published on October 2, 2006 as Manuscript M600320-MCP200 Improved Validation of Peptide MS/MS Assignments Using Spectral Intensity Prediction Shaojun Sun 1, Karen Meyer-Arendt 2,
Translational Biomarker Core
 Translational Biomarker Core Instrumentation Thermo Scientific TSQ Quantum Triple Quadrupole Mass Spectrometers. There are two TSQ Quantum Ultra AM instruments available in the TBC. The TSQ Quantum Ultra
Translational Biomarker Core Instrumentation Thermo Scientific TSQ Quantum Triple Quadrupole Mass Spectrometers. There are two TSQ Quantum Ultra AM instruments available in the TBC. The TSQ Quantum Ultra
The Pitfalls of Peaklist Generation Software Performance on Database Searches
 Proceedings of the 56th ASMS Conference on Mass Spectrometry and Allied Topics, Denver, CO, June 1-5, 2008 The Pitfalls of Peaklist Generation Software Performance on Database Searches Aenoch J. Lynn,
Proceedings of the 56th ASMS Conference on Mass Spectrometry and Allied Topics, Denver, CO, June 1-5, 2008 The Pitfalls of Peaklist Generation Software Performance on Database Searches Aenoch J. Lynn,
MassHunter Software Overview
 MassHunter Software Overview 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks A
MassHunter Software Overview 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks A
Yifei Bao. Beatrix. Manor Askenazi
 Detection and Correction of Interference in MS1 Quantitation of Peptides Using their Isotope Distributions Yifei Bao Department of Computer Science Stevens Institute of Technology Beatrix Ueberheide Department
Detection and Correction of Interference in MS1 Quantitation of Peptides Using their Isotope Distributions Yifei Bao Department of Computer Science Stevens Institute of Technology Beatrix Ueberheide Department
QTOF-based proteomics and metabolomics for the agro-food chain.
 QTOF-based proteomics and metabolomics for the agro-food chain luigi.lucini@unicatt.it Metabolomics Two scenarios identification of known unknowns and unknown unknowns For known unknowns use spectral or
QTOF-based proteomics and metabolomics for the agro-food chain luigi.lucini@unicatt.it Metabolomics Two scenarios identification of known unknowns and unknown unknowns For known unknowns use spectral or
Tutorial 1: Setting up your Skyline document
 Tutorial 1: Setting up your Skyline document Caution! For using Skyline the number formats of your computer have to be set to English (United States). Open the Control Panel Clock, Language, and Region
Tutorial 1: Setting up your Skyline document Caution! For using Skyline the number formats of your computer have to be set to English (United States). Open the Control Panel Clock, Language, and Region
Overview. Introduction. André Schreiber AB SCIEX Concord, Ontario (Canada)
") Quantitation and Identification of Pharmaceuticals and Personal Care Products (PPCP) in Environmental Samples using Advanced TripleTOF MS/MS Technology André Schreiber AB SCIEX Concord, Ontario (Canada)
Quantitation and Identification of Pharmaceuticals and Personal Care Products (PPCP) in Environmental Samples using Advanced TripleTOF MS/MS Technology André Schreiber AB SCIEX Concord, Ontario (Canada)
Reagents. Affinity Tag (Biotin) Acid Cleavage Site. Figure 1. Cleavable ICAT Reagent Structure.
 Acid Cleavage Site. Figure 1. Cleavable ICAT Reagent Structure.") DATA SHEET Protein Expression Analysis Reagents Background The ultimate goal of proteomics is to identify and quantify proteins that are relevant to a given biological state; and to unearth networks of
DATA SHEET Protein Expression Analysis Reagents Background The ultimate goal of proteomics is to identify and quantify proteins that are relevant to a given biological state; and to unearth networks of
(Refer Slide Time 00:09) (Refer Slide Time 00:13)
 (Refer Slide Time 00:13)") (Refer Slide Time 00:09) Mass Spectrometry Based Proteomics Professor Sanjeeva Srivastava Department of Biosciences and Bioengineering Indian Institute of Technology, Bombay Mod 02 Lecture Number 09 (Refer
(Refer Slide Time 00:09) Mass Spectrometry Based Proteomics Professor Sanjeeva Srivastava Department of Biosciences and Bioengineering Indian Institute of Technology, Bombay Mod 02 Lecture Number 09 (Refer
Novel quadrupole time-of-flight mass spectrometry for shotgun proteomics
 DISSERTATION ZUR ERLANGUNG DES DOKTORGRADES DER FAKULTÄT FÜR CHEMIE UND PHARMAZIE DER LUDWIG-MAXIMILIANS-UNIVERSITÄT MÜNCHEN Novel quadrupole time-of-flight mass spectrometry for shotgun proteomics von
DISSERTATION ZUR ERLANGUNG DES DOKTORGRADES DER FAKULTÄT FÜR CHEMIE UND PHARMAZIE DER LUDWIG-MAXIMILIANS-UNIVERSITÄT MÜNCHEN Novel quadrupole time-of-flight mass spectrometry for shotgun proteomics von
Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis
 Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis Technical Overview Introduction Metabolomics studies measure the relative abundance of metabolites
Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis Technical Overview Introduction Metabolomics studies measure the relative abundance of metabolites
SILAC and TMT. IDeA National Resource for Proteomics Workshop for Graduate Students and Post-docs Renny Lan 5/18/2017
 SILAC and TMT IDeA National Resource for Proteomics Workshop for Graduate Students and Post-docs Renny Lan 5/18/2017 UHPLC peak chosen at 26.47 min LC Mass at 571.36 chosen for MS/MS MS/MS MS This is a
SILAC and TMT IDeA National Resource for Proteomics Workshop for Graduate Students and Post-docs Renny Lan 5/18/2017 UHPLC peak chosen at 26.47 min LC Mass at 571.36 chosen for MS/MS MS/MS MS This is a
Targeted protein quantification
 Targeted Quantitative Proteomics Targeted protein quantification with high-resolution, accurate-mass MS Highly selective Very sensitive Complex samples HR/AM A more complete quantitative proteomics picture
Targeted Quantitative Proteomics Targeted protein quantification with high-resolution, accurate-mass MS Highly selective Very sensitive Complex samples HR/AM A more complete quantitative proteomics picture
LogViewer: A Software Tool to Visualize Quality Control Parameters to Optimize Proteomics Experiments using Orbitrap and LTQ-FT Mass Spectrometers
 COMMUNICATION LogViewer: A Software Tool to Visualize Quality Control Parameters to Optimize Proteomics Experiments using Orbitrap and LTQ-FT Mass Spectrometers Michael J. Sweredoski, Geoffrey T. Smith,
COMMUNICATION LogViewer: A Software Tool to Visualize Quality Control Parameters to Optimize Proteomics Experiments using Orbitrap and LTQ-FT Mass Spectrometers Michael J. Sweredoski, Geoffrey T. Smith,
Genome wide analysis of protein and mrna half lives reveals dynamic properties of mammalian gene expression
 Genome wide analysis of protein and mrna half lives reveals dynamic properties of mammalian gene expression Matthias Selbach Cell Signaling and Mass Spectrometry Max Delbrück Center for Molecular Medicine
Genome wide analysis of protein and mrna half lives reveals dynamic properties of mammalian gene expression Matthias Selbach Cell Signaling and Mass Spectrometry Max Delbrück Center for Molecular Medicine
MSc Chemistry Analytical Sciences. Advances in Data Dependent and Data Independent Acquisition for data analysis in proteomic research
 MSc Chemistry Analytical Sciences Literature Thesis Advances in Data Dependent and Data Independent Acquisition for data analysis in proteomic research by Florian L. R. Lucas 11198877 September 2016 12
MSc Chemistry Analytical Sciences Literature Thesis Advances in Data Dependent and Data Independent Acquisition for data analysis in proteomic research by Florian L. R. Lucas 11198877 September 2016 12
Tandem mass spectra were extracted from the Xcalibur data system format. (.RAW) and charge state assignment was performed using in house software
 and charge state assignment was performed using in house software") Supplementary Methods Software Interpretation of Tandem mass spectra Tandem mass spectra were extracted from the Xcalibur data system format (.RAW) and charge state assignment was performed using in house
Supplementary Methods Software Interpretation of Tandem mass spectra Tandem mass spectra were extracted from the Xcalibur data system format (.RAW) and charge state assignment was performed using in house
Nontarget Analysis via LC-QTOF-MS to Assess the Release of Organic Substances from Polyurethane Coating
 Nontarget Analysis via LC-QTOF-MS to Assess the Release of Organic Substances from Polyurethane Coating Agnessa Luft, Kathrin Bröder, Uwe Kunkel,#, Manoj Schulz, Christian Dietrich, Roland Baier, Peter
Nontarget Analysis via LC-QTOF-MS to Assess the Release of Organic Substances from Polyurethane Coating Agnessa Luft, Kathrin Bröder, Uwe Kunkel,#, Manoj Schulz, Christian Dietrich, Roland Baier, Peter
CSE182-L8. Mass Spectrometry
 CSE182-L8 Mass Spectrometry Project Notes Implement a few tools for proteomics C1:11/2/04 Answer MS questions to get started, select project partner, select a project. C2:11/15/04 (All but web-team) Plan
CSE182-L8 Mass Spectrometry Project Notes Implement a few tools for proteomics C1:11/2/04 Answer MS questions to get started, select project partner, select a project. C2:11/15/04 (All but web-team) Plan
MALDI-HDMS E : A Novel Data Independent Acquisition Method for the Enhanced Analysis of 2D-Gel Tryptic Peptide Digests
 -HDMS E : A Novel Data Independent Acquisition Method for the Enhanced Analysis of 2D-Gel Tryptic Peptide Digests Emmanuelle Claude, 1 Mark Towers, 1 and Rachel Craven 2 1 Waters Corporation, Manchester,
-HDMS E : A Novel Data Independent Acquisition Method for the Enhanced Analysis of 2D-Gel Tryptic Peptide Digests Emmanuelle Claude, 1 Mark Towers, 1 and Rachel Craven 2 1 Waters Corporation, Manchester,
MS-MS Analysis Programs
 MS-MS Analysis Programs Basic Process Genome - Gives AA sequences of proteins Use this to predict spectra Compare data to prediction Determine degree of correctness Make assignment Did we see the protein?
MS-MS Analysis Programs Basic Process Genome - Gives AA sequences of proteins Use this to predict spectra Compare data to prediction Determine degree of correctness Make assignment Did we see the protein?
Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry
 Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry by Xi Han A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree
Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry by Xi Han A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree
Statistical analysis of isobaric-labeled mass spectrometry data
 Statistical analysis of isobaric-labeled mass spectrometry data Farhad Shakeri July 3, 2018 Core Unit for Bioinformatics Analyses Institute for Genomic Statistics and Bioinformatics University Hospital
Statistical analysis of isobaric-labeled mass spectrometry data Farhad Shakeri July 3, 2018 Core Unit for Bioinformatics Analyses Institute for Genomic Statistics and Bioinformatics University Hospital
Applications of Mass Spectrometry for Biotherapeutic Characterization
 Applications of Mass Spectrometry for Biotherapeutic Characterization Case Studies of Disulfide Characterization and Separation free Modes of Analysis Steven L. Cockrill Amgen Colorado Analytical Sciences
Applications of Mass Spectrometry for Biotherapeutic Characterization Case Studies of Disulfide Characterization and Separation free Modes of Analysis Steven L. Cockrill Amgen Colorado Analytical Sciences
Pushbutton Units and Indicator Lights
 Insert labels and insert caps Clear, illuminated and indicator lights can be fitted with insert labels and caps for identification purposes. These labels and caps are made of a semi-transparent molded
Insert labels and insert caps Clear, illuminated and indicator lights can be fitted with insert labels and caps for identification purposes. These labels and caps are made of a semi-transparent molded
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra. Andrew Keller
 PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
DeMix Workflow for Efficient Identification of Co-fragmented. Peptides in High Resolution Data-dependent Tandem Mass
 MCP Papers in Press. Published on August 6, 2014 as Manuscript O114.038877 DeMix Workflow for Efficient Identification of Co-fragmented Peptides in High Resolution Data-dependent Tandem Mass Spectrometry
MCP Papers in Press. Published on August 6, 2014 as Manuscript O114.038877 DeMix Workflow for Efficient Identification of Co-fragmented Peptides in High Resolution Data-dependent Tandem Mass Spectrometry
Spectrum-to-Spectrum Searching Using a. Proteome-wide Spectral Library
 MCP Papers in Press. Published on April 30, 2011 as Manuscript M111.007666 Spectrum-to-Spectrum Searching Using a Proteome-wide Spectral Library Chia-Yu Yen, Stephane Houel, Natalie G. Ahn, and William
MCP Papers in Press. Published on April 30, 2011 as Manuscript M111.007666 Spectrum-to-Spectrum Searching Using a Proteome-wide Spectral Library Chia-Yu Yen, Stephane Houel, Natalie G. Ahn, and William
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra
 PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Day 2 October 17, 2006 Andrew Keller Rosetta Bioinformatics, Seattle Outline Need to validate peptide assignments to MS/MS
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Day 2 October 17, 2006 Andrew Keller Rosetta Bioinformatics, Seattle Outline Need to validate peptide assignments to MS/MS
TOMAHAQ Method Construction
 TOMAHAQ Method Construction Triggered by offset mass accurate-mass high-resolution accurate quantitation (TOMAHAQ) can be performed in the standard method editor of the instrument, without modifications
TOMAHAQ Method Construction Triggered by offset mass accurate-mass high-resolution accurate quantitation (TOMAHAQ) can be performed in the standard method editor of the instrument, without modifications
Screening of pesticides residues by the time of flight analyzer (ToF) : Myth or reality?
 : Myth or reality?") Screening of pesticides residues by the time of flight analyzer (ToF) : Myth or reality? Laure Joly & Vincent Hanot WIV-ISP, Juliette Wytsmanstraat 14, 15 Brussel The passengers pesticides are requested......
Screening of pesticides residues by the time of flight analyzer (ToF) : Myth or reality? Laure Joly & Vincent Hanot WIV-ISP, Juliette Wytsmanstraat 14, 15 Brussel The passengers pesticides are requested......
Nature Methods: doi: /nmeth Supplementary Figure 1. Fragment indexing allows efficient spectra similarity comparisons.
 Supplementary Figure 1 Fragment indexing allows efficient spectra similarity comparisons. The cost and efficiency of spectra similarity calculations can be approximated by the number of fragment comparisons
Supplementary Figure 1 Fragment indexing allows efficient spectra similarity comparisons. The cost and efficiency of spectra similarity calculations can be approximated by the number of fragment comparisons
Developing Algorithms for the Determination of Relative Abundances of Peptides from LC/MS Data
 Developing Algorithms for the Determination of Relative Abundances of Peptides from LC/MS Data RIPS Team Jake Marcus (Project Manager) Anne Eaton Melanie Kanter Aru Ray Faculty Mentors Shawn Cokus Matteo
Developing Algorithms for the Determination of Relative Abundances of Peptides from LC/MS Data RIPS Team Jake Marcus (Project Manager) Anne Eaton Melanie Kanter Aru Ray Faculty Mentors Shawn Cokus Matteo
Protein Sequencing Research Group ABRF 2015 annual meeting
 Protein Sequencing Research Group ABRF 2015 annual meeting » N-terminal sequencing is in the midst of a technology transition from classical Edman sequencing to mass spectrometry (MS)-based sequencing»
Protein Sequencing Research Group ABRF 2015 annual meeting » N-terminal sequencing is in the midst of a technology transition from classical Edman sequencing to mass spectrometry (MS)-based sequencing»
Your mass spec s sidekick
 ZipChip Give your mass spec some zip Your mass spec s sidekick The ZipChip platform prepares and separates a wide range of biological samples, then electrosprays them into your mass spec for analysis.
ZipChip Give your mass spec some zip Your mass spec s sidekick The ZipChip platform prepares and separates a wide range of biological samples, then electrosprays them into your mass spec for analysis.