arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 6 Apr 2000
|
|
|
- Magdalene Summers
- 6 years ago
- Views:
Transcription
1 Electronic Structure and Magnetism of Equiatomic FeN Y. Kong Department of Physics & The Applied Magnetics Laboratory of the Ministry of Education, Lanzhou University, 73 Lanzhou, China Max-Planck-Institut für Festkörperforschung, Heisenbergstr., D-7569 Stuttgart, Germany arxiv:cond-mat/49v [cond-mat.mtrl-sci] 6 Apr 2 Abstract In order to investigate the phase stability of equiatomic FeN compounds and the structuredependent magnetic properties, the electronic structure and total energy of FeN with NaCl, ZnS and CsCl structures and various magnetic configurations are calculated using the firstprinciple TB-LMTO-ASA method. Among all the FeN phases considered, the antiferromagnetic (AFM) NaCl structure with q = (,, π) is found to have the lowest energy at the theoretical equilibrium volume. However, the ferromagnetic (FM) NaCl phase lies only mry higher. The estimated equilibrium lattice constant a th =4.36Å for nonmagnetic (NM) ZnS-type FeN agrees quite well with the experimental value of a exp =4.33Å but for AFM NaCl phase the a th =4.2Å is 6.7% smaller than the value observed experimentally. For ZnS-type FeN, metastable magnetic states are found for volumes larger than the equilibrium value. With the analysis of atom- and orbital-projected density of states (DOS) and orbital-resolved crystal orbital Hamilton population (COHP) the iron-nitrogen interactions in NM-ZnS, AFM-NaCl and FM-CsCl structures are discussed. The leading Fe-N interaction is due to the d-p iron-nitrogen hybridization while considerable s-p and p-p hybridizations are also observed in all three phases. The iron magnetic moment µ Fe in FeN is found to be highly sensitive to the nearest-neighboring Fe-N distance. In particular, the µ Fe in ZnS and CsCl structures show an abrupt drop from the value of about 2µ B to zero with the reduction of the Fe-N distance. Keywords: FeN, TB-LMTO-ASA, magnetism, electronic structure
2 Introduction For a long time iron nitride has attracted much scientific interest in basic research as well as in technology-oriented research. While a large number of results for iron-rich nitrides, such as γ -Fe 4 N, have appeared in the literature[], only a few investigations on high N- content FeN, specifically FeN[2, 3] with the proportionality : of Fe and N atoms were reported. Recently, two kinds of FeN structures have been determined in FeN films[4, 5, 6] synthesized by sputtering technique. They are the sodium chloride structure with lattice constants a=4.5å and the zinc blende structure with lattice constant a 4.33Å. All the prepared FeN films were non-stoichiometric due to N vacancies and/or impurities. 57 Fe Mössbauer spectroscopy experiments[4, 7] have shown that at 4.2K no magnetic hyperfine splitting is observed for ZnS-type FeN, but two kinds of Fe sites in NaCl-type FeN exhibit surprisingly large hyperfine fields of 49T and 3T. It was suggested that the ZnS-type FeN is nonmagnetic (NM) while the NaCl-type FeN shows antiferromagnetic (AFM) coupling[4, 7]. However, the magnetic measurements by Suzuki et al.[5] indicated that at low temperature the stoichiometric ZnS-type FeN is AFM and exhibits a micromagnetic character. Using a full-potential linearized-augmented-plane-wave (FLAPW) method, Shimizu et al.[8, 9] calculated the electronic, structural and magnetic properties of stoichiometric ZnSand NaCl-type FeN and estimated the equilibrium lattice constants and bulk moduli. The results indicated that the ferromagnetic (FM) NaCl-type FeN was more stable than the NM-ZnS structure. Furthermore, the calculated results[9] for the NaCl-type FeN with NM, FM, (π, π, π)- and (,, π)-afm configurations identified that the FM structure was the ground state with equilibrium lattice constant a th 4.Å. It has further shown that at the experimental lattice constant 4.5Å the (π, π, π)-afm state was more stable than the FM state. A similar detailed analysis was not carried out for the ZnS-type FeN and the reason for the difference in magnetism between the two structures was therefore not clear. Most recently, Eck et al.[] performed band structure calculations using tight-binding-linearmuffin-tin-orbitals (TB-LMTO) method within the atomic-sphere-approximation (ASA) and investigated structural and electronic properties of both FeN structures by analyzing density of states (DOS) and crystal orbital Hamilton population (COHP). They concluded that the zinc blende structure should be more stable because of the weaker antibonding Fe-Fe interactions below the Fermi level. Moreover, the authors evaluated the theoretical lattice constants for non-stoichiometric FeN and found that the deficiency of N atom decreases the equilibrium lattice constant. The magnetic properties of FeN were however hardly discussed. To achieve a better understanding of the magnetic diversity in FeN, systematic investigation 2
3 of the electronic structure and magnetic properties of various FeN structures thus seems to be needed. In this paper, the TB-LMTO-ASA method is applied to calculate band structures of FeN compounds with various structures and spin configurations. The structural, electronic and magnetic properties of the FeN are thus investigated by analyzing the calculated electronic structure and total energy. In addition to the NaCl and ZnS structures, we also consider FeN in the CsCl structure. A brief description of computational technique will be given in the next section. In section 3, we present the calculated total energy and electronic structural and discuss the structure and magnetic properties of FeN compounds. A short summary is given in the last section. 2 Computational Details In the present study the electronic structures of equiatomic FeN compounds are calculated self-consistently using the scalar-relativistic TB-LMTO method[2] in the atomicsphere-approximation including the combined correction. A local-density-approximation (LDA)[3] to the density-functional theory (DFT) is applied and the non-local correction is also included using the Perdew-Wang generalized-gradient-approximation (GGA).[4] The lattice structures of NaCl-, ZnS- and CsCl-type FeN compounds are illustrated in Fig.. The Fe atoms in sodium chloride and zinc blende structures form an fcc lattice with different coordination of the N atoms. In the NaCl structure the N atoms are located at octahedral sites of the fcc lattice and each Fe atom has six N neighbors while in the ZnS structure the half-filling of the N atoms in tetrahedral sites of the fcc lattice makes each Fe four-fold coordinated to N atoms. In the cesium chloride structure the N atoms fill the body-centered sites of a simple-cubic Fe lattice and one Fe is eight-fold coordinated to N. The sites and filling fractions of the N atoms, the experimental magnetic structure, the nearest-neighbor (NN) Fe-Fe and Fe-N distances as well as the number of N neighbors of an Fe atom are summarized for the different structures in Table. We perform NM, FM and (,, π)-afm calculations for FeN with NaCl, ZnS and CsCl structures. Besides (,, π)-afm state, the (π, π, π)-afm configuration and an additional AFM structure[], which consists of double layers with FM interlayer coupling along []- direction, AFMD, are also considered for NaCl-type FeN to include possible magnetic coupling in FeN film. The k-space integration is performed with the tetrahedron method[5] using mesh within the Brillouin zone. All partial waves with l 2 are included in the basis for Fe as well as N. For ZnS- and NaCl-type FeN, special care is taken in filling 3
4 the interatomic space since the structures are rather open. Therefore, it is necessary to introduce interstitial spheres. The sphere radii and the positions of interstitial spheres are chosen in such a way that space filling is achieved without exceeding a sphere overlap of 6%. This is done using an automatic procedure developed by Krier et al.[6]. The radii of atomic and interstitial spheres corresponding to the theoretical equilibrium volume are listed in Table 2 for the different FeN structures. In combination with the analysis of atom- and orbital-projected density of states (DOS), the COHP technique[7] is applied to analyze the chemical bonding in ZnS-, NaCl- and CsCltype FeN to examine the different Fe-N interactions. This technique provides information analogous to the Crystal Orbital Overlap Population (COOP) analysis.[8] While COOP curves are energy resolved plots of the Mulliken overlap population between two atoms or orbitals, a COHP curve is an energy resolved plot of the contribution of a given bond to the bonding energy of the system. Similar to COOP curves, in all the COHP curves presented here positive values are bonding and negative antibonding, i.e. COHP is plotted instead of COHP. 3 Results and Discussions 3. Total energy and phase stability The calculated total-energy curves for ZnS-, NaCl- and CsCl-type FeN with NM, FM and AFM spin configurations are shown in Fig. 2. The theoretical equilibrium lattice constants a th are estimated for various FeN phases and they are listed in Table 2 together with total energies at a th. The total-energy for NM CsCl structure is not plotted in Fig. 2 because the values are much higher than the other phases. Firstly, we shall discuss the total-energy curves for NaCl-type FeN. Except for the NM NaCl-type FeN with higher energy, the magnetic NaCl phases with different spin configurations give similar total-energy curves. As indicated in Table 2, at the equilibrium volume the E th for (,, π)-afm phase is only mry lower than that for FM-NaCl structure and mry below the highest (π, π, π)-afm structure. Although our results are in better agreement with experiment than previous results[9], the a th ( 4.2Å) estimated for NaCl-type FeN is still 6.7% smaller than the values observed experimentally. This discrepancy is much larger than the normal deviation due to the local density approximation (LDA) with GGA correction. It may therefore be, as suggested in Refs.[9, ], that the experimental NaCltype FeN with a=4.5å is not in stable state due to unknown effects, such as the effect of the surface. We also perform total-energy calculations for magnetic NaCl phases without the inclusion 4
5 of the GGA correction to the LDA. The estimated theoretical lattice constants for magnetic NaCl phases are about Å, which are smaller than those obtained with GGA, and the FM phase is found to be stable at the theoretical equilibrium volume. The results are consistent with those by Shimizu et al.[9]. In the following only the results calculated with the GGA correction are presented. For ZnS-type FeN, a NM ground state is found with theoretical equilibrium lattice constant a th =4.36Å, which agrees quite well with the experimental value[4]. The state is about 6mRy higher in energy than that of the (,, π)-afm NaCl structure. Self-consistent (,, π)-afm and FM solutions are found when the lattice constant is larger than 4.45Å and 4.69Å, respectively. Since the critical lattice constant 4.45Å for the (,, π)-afm state is only.9å larger than a th of the NM-ZnS phase, one could expect that the metastable AFM phase would be important for the observed micromagnetic character in ZnS-type FeN.[5] The total-energy curves calculated for FM and (,, π)-afm CsCl-type FeN are nearly identical and about 7mRy above those of ZnS and NaCl structures. Because of this rather higher total-energy, the CsCl-structure should not be considered a possible stable FeN phase. Among all the investigated FeN phases, as mentioned above, the simple (,, π)-afm NaCl structure is found to have the lowest total energy at theoretical equilibrium volume, while the E th for FM-NaCl structure is only about mry higher. However, the energy difference is very small and may be of the order of the inaccuracies due to the ASA used in our calculations. 3.2 Electronic structure and iron-nitrogen interactions In the following, the electronic structure and Fe-N interactions in the FeN phases will be analyzed in detail using the calculated DOS and COHP for NM ZnS, (,, π)-afm NaCl and FM CsCl structures. In Fig. 3 we show the spin-resolved DOS for NM ZnS-, (,, π)-afm NaCl- and FM CsCl-type FeN at the theoretical equilibrium volume. The DOS at Fermi level, N(E F ), are listed in Table 2. Except for the s-like states around 5eV, which are not shown, the DOS of NM-ZnS structure shown in Fig. 3a mainly consists of two sets of structures, which are separated by a 2eV energy gap. The lower energy set, centered around 6eV, is primarily composed of N 2p bands with some admixture of Fe d character, the higher energy set from 2.3 to 3.6eV, is dominated by Fe 3d states. A small gap at.5ev further divides the latter into two parts, which are respectively characterized by the crystal-field splitted e and t 2 sets. The Fermi-level E F is located just above the gap at the low-energy side of the t 2 set. The reason for ZnS-type FeN phase showing no magnetic moment at equilibrium volume may be 5
6 understood from the DOS at the E F. According to the Stoner model the self-consistency condition for a ferromagnet can be simply expressed by N(E F ) I = where N(E F ) is the paramagnetic density of state at the E F and I the effective Stoner interaction parameter. For the ZnS-type FeN, it is observed that the DOS curve intersects the Fermi-level with a large negative slope and gives a quite small N(E F ) (.7 states/ev spin). Consequently, no magnetic ordering is observed for ZnS-type FeN. On the other hand, by increasing the volume, the 3d subband becomes much narrower and the N(E F ) larger. The Stoner criterion may therefore be satisfied and the ZnS-type FeN shows certain magnetic ordering. Indeed, our total-energy calculations for ZnS-type FeN has confirmed the metastable FM and AFM phases existing at larger volumes. Since the band structures of two sublattice in AFM phase are identical, in Fig. 3b only the sublattice DOS for (,, π)-afm NaCl structure is plotted. Compared to that of NM ZnS phase, the DOS of (,, π)-afm NaCl-type FeN form a continuous spectrum over the energy range between 8 and 4eV. Due to the large dispersion of Fe 3d subbands the expected crystal field splitting of 3d orbitals can not be clearly seen. While the majority-spin DOS are nearly completely occupied, a dominant minority-spin peak is located above the Fermi level due to the exchange-splitting. In Fig. 3c the peaks of the DOS for FM CsCl structure also show complicated structure. Compared to that of NaCl structure, the DOS of the CsCl-type FeN exhibits larger dispersion. Below the E F, the majority-spin DOS peaks of NaCl and CsCl structures have a tendency to split into two parts by a deep valley at about 3 and 4eV, respectively. The higher part is dominated by Fe 3d states while the lower one is mostly composed of N 2p with some admixture of Fe d states. The partial DOS (PDOS) of FeN with NM ZnS, (,, π)-afm NaCl and FM CsCl structures, projected for Fe s, p, d and N p orbitals, are plotted in Fig. 4. Correspondingly, Fig. 5 shows the orbital-projected COHP calculated for the s-p, p-p and d-p iron-nitrogen interactions. Here only the nearest-neighbor contributions are included in the COHP. Noticed that of the two identical sublattices in (,, π)-afm-nacl phase, only the PDOS for one sublattice are shown in Fig. 4b while the COHP curves for Fe -N (Fe 2 -N 2 ) intra-sublattice and Fe -N 2 (Fe 2 -N ) inter-sublattice interactions are presented in Fig. 5b and 5c, respectively. Here Fe (N ) defines the Fe (N) atom in the up-polarized sublattice and Fe 2 (N 2 ) the atom in the down-polarized sublattice. Consistent with the observation of the total DOS, a 2eV energy gap separates the PDOS of NM ZnS phase, shown in Fig. 4a, into two sets of isolated structures. As indicated by the COHP curves in Fig. 5a, N p orbitals mix Fe s, p and d subbands on both side of 6
7 the gap but the d-p hybridization dominates the iron-nitrogen interactions. The d-p Fe-N hybridization is characterized by the large bonding peaks below the gap, centered at about 5eV, and the antibonding peaks around the Fermi-level, centered at about 2.eV. On the contrary, in both energy windows the s-p and p-p hybridizations form bonding interactions. The corresponding antibonding interactions lie far above the Fermi-level. In (,, π)-afm NaCl structure, the PDOS projected for Fe s, p, d and N p orbitals (Fig. 4b) spread over the same energy range from 8 to 4.eV without energy gap and show prominent hybridizations in the entire energy range. According to the calculated COHP shown in Fig. 5b and Fig. 5c, the d-p hybridization in NaCl-type FeN is characteristic of the bonding interactions below 3eV and the antibonding states above 3eV. Furthermore, it is found that the antibonding peaks below the E F are dominated by antibonding π states and the peaks above the E F are mainly σ states in character. Comparing the d-p interactions in Fig. 5b and Fig. 5c, a stronger antibonding π peak at 2eV is observed for inter-sublattice Fe-N hybridization. Except for the leading d-p interactions the s-p and p-p hybridization show considerable bonding interactions from 8 to 4eV. In contrast to the d-p interaction, the s-p and p-p interactions for both intra- and inter-sublattice Fe-N interactions are nearly identical. Compared to the ZnS structure, the antibonding peaks of s-p and p-p hybridization in NaCl structure are shifted towards lower energy. Due to larger Fe-N distance, the Fe-N interactions in FM CsCl structure is much weaker than that in ZnS and NaCl structure. As also observed in (,, π)-afm NaCl structure, the dominant Fe-N d-p hybridization is composed of the bonding set at lower energy and the antibonding set around the E F. Below the E F there exist strong antibonding peaks. For s-p and p-p interactions the antibonding peaks are found to be much lower in energy than those in ZnS and NaCl structures. Although the s-p and p-p hybridizations give inevitable contributions, as indicated by the COHP curves in Fig. 5, the dominant Fe-N interaction in all the three FeN structures is the d-p hybridization. In Fig. 5 we also plot the integrated COHP for d-p interactions. As indicated by the ICOHP at Fermi level, the ZnS-type FeN exhibits the strongest d-p Fe-N hybridization among the three FeN structures because of the shortest nearest-neighboring Fe-N distance. On the contrary, the largest Fe-N distance in CsCl structure makes the CsCltype FeN showing the weakest Fe-N hybridization although the Fe atoms in this structure has the most N neighbors. 7
8 3.3 Magnetic moment and its volume dependence The calculated iron magnetic moments µ Fe for the different FeN phases at the theoretical equilibrium volume are listed in table 2. For all the four magnetic NaCl-type FeN phases a similar value of µ Fe is obtained. Among them the FM phase produces a slightly larger µ Fe. For both FM and AFM CsCl-type FeN the calculated Fe magnetic moments are larger than those in NaCl-type FeN. At equilibrium volume no spin-splitting for FeN with ZnS structure is obtained from spin-polarized FM and AFM calculations. Noticed that the Fe-N interaction in ZnS structure is the strongest and that in CsCl structure the weakest, it is reasonably concluded that the magnetic properties of Fe in FeN structures is strongly correlated to the strength of the Fe-N interactions, mainly d-p hybridization. In other words, the magnetic properties of FeN phases is closely related to the nearest-neighboring Fe-N distance in the structure. The stronger the Fe-N hybridization is, the smaller becomes the Fe magnetic moment. The Fe magnetic moments in magnetic NaCl-, CsCl- and ZnS-type FeN calculated at various volumes are shown in Fig. 6 as a function of the nearest-neighboring Fe-N distance, d Fe N. It is found that the µ Fe is highly sensitive to the d Fe N. By compressing the volume, the Fe magnetic moment in all the magnetic FeN phases dramatically decrease with the reduction of the d Fe N. While µ Fe in NaCl-type FeN decrease with d Fe N at nearly the same rate and it becomes zero gradually when the d Fe N is smaller than about 3.3Å, µ Fe in both CsCl and ZnS structures exhibit a sudden drop from a value of about 2µ B to zero at a certain critical d Fe N. According to the canonical band model[9], the different volume dependence of the µ Fe in various FeN structures should be related to the difference of the DOS around the E F. 4 Conclusions Starting out from the calculated total energy and electronic structure using the TB- LMTO-ASA method, we have investigated structural, electronic and magnetic properties of FeN compounds with NaCl, ZnS and CsCl structures. From the calculated total-energy a stable (,, π)-afm NaCl-type FeN with theoretical equilibrium lattice constant a = 4.2Å is identified. For the FeN with ZnS structure, our results indicate the existence of metastable AFM and FM solutions with a larger cell volume besides stable NM phase. These metastable states may be important for the observed micromagnetic character in ZnS-type FeN. By analyzing the atom- and orbital-resolved density of states and the orbital-projected 8
9 COHP, the Fe-N hybridization interactions in NM ZnS, (,, π)-afm NaCl and FM CsCl structures are discussed in detail. In all the three considered FeN structures the d-p hybridization between Fe and N atoms dominates the Fe-N interactions. With the calculated Fe magnetic moment for various FeN structures, it is suggested that the magnetic properties of FeN phases are closely related to the strength of the Fe-N d-p hybridization. Furthermore, the Fe magnetic moment in FeN phases are found to be highly sensitive to the nearest-neighboring Fe-N distance. Finally, it should be mentioned that the equilibrium lattice constant a th estimated for NaCl-type FeN is 6.7% smaller than the values observed experimentally though in our calculations the GGA correction to the local density approximation has been applied. It is suggested that other unknown effects should be responsible for the extraordinarily larger lattice constant observed for FeN with NaCl structure. Eck et al.[] have estimated the equilibrium lattice constant for defected FeN and obtained a rather smaller a th. Up to date, no theoretical investigations on FeN film has been reported. It is highly desired to perform calculations on FeN structure with low-dimensional symmetry so that the effect of surface may be examined. Acknowledgements The author would like to gratefully acknowledge Prof. O.K. Andersen for many helpful advices and is indebted to Dr. O. Jepsen for a careful reading of the manuscript. 9
10 References [] P. Mohn and S.F. Matar, J. Magn. Magn. Mater., 9, 234(999), and the references listed therein. [2] N. Heiman and N.S. Kazama, J. Appl. Phys., 52, 3562(98). [3] A. Oueldennaoua, E. Bauer-Grosse, M. Foos and C. Frants, Scr. Metall., 9, 53(985). [4] H. Nakagawa, S. Nasu, H. Fujii, M. Takahashi and F. Kanamaru, Hyper. Inter., 69, 455(99). [5] K. Suzuki, H. Morita, T. Kaneko, H. Yoshida and H. Fujimori, J. Alloys & Compounds, 2, (993). [6] L. Rissanen, M. Neubauer, K.P. Lieb, P. Schaaf, J. Alloys & Compounds, 274, 74(998). [7] T. Hinomura and S. Nasu, Hyper. Inter.,, 22(998). [8] H. Shimizu, M. Shirai and N. Suzuki, J. Phys. Soc. Jap., 66, 347(997). [9] H. Shimizu, M. Shirai and N. Suzuki, J. Phys. Soc. Jap., 67, 922(998). [] B. Eck, R. Dronskowski, M. Takahashi and S. Kikkawa, J. Mater. Chem., 9, 527(999). [] H.C. Herper, E. Hoffmann and P. Entel, Phys. Rev. B 6, 3839(999). [2] O.K. Andersen and O. Jepsen, Phys. Rev. Lett. 53, 257(984); G. Krier, O. Jepsen, A. Burkhardt and O.K. Andersen, The Stuttgart TB-LMTO-ASA program. [3] S.H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 58, 2(98). [4] J.P. Perdew and Y. Wang, Phys. Rev. B 45, 3244(992). [5] O. Jepsen and O.K. Andersen, Solid State Commun. 9, 763(97) and Phys. Rev. B 29, 5965(984); P. Blöchl, O. Jepsen and O.K. Andersen, Phys. Rev. B 49, 6223(994). [6] G. Krier, O.K. Andersen, O. Jepsen (unpublished). [7] R. Dronskowski and P.E. Blöchl, J. Phys. Chem., 97, 867(993). [8] T. Hughbanks and R. Hoffmann, J. Am. Chem. Soc., 5, 3528(983). [9] O.K. Andersen, J. Madsen, U.K. Poulsen, O. Jepsen and J. Kollar, Physica, 86-88B, 249(977).
11 Tables Table : The site and filling fraction (per cubic cell) of the N atoms in FeN compounds, lattice constant a and magnetic structures. d Fe Fe and d Fe N are the nearest-neighbor Fe-Fe and Fe-N distances at the experimental lattice constant. n is the number of N neighbors of one Fe atom. Since the CsCl-type FeN does not exist, the calculated results are given. Structure type ZnS NaCl CsCl Site tetrahedral octahedral body-centered filling fraction 4/8 4/4 / exp. mag. structure NM AFM FM a (Å) 4.33[4] 4.5[4] 2.63 d Fe Fe (Å) d Fe N (Å) n Table 2: Calculated total energy, equilibrium lattice constant, DOS at E F and Fe magnetic moment for some considered FeN structures. E th is the total energy (given by E in Ry/FeN) at the theoretical equilibrium lattice constants a th, N(E F ) the DOS at Fermi level in states/ev FeN and µ Fe the Fe magnetic moment at the a th. S Fe and S N are the radii of Fe and N atomic spheres corresponding to the theoretical equilibrium volume and S E the radius of interstitial sphere. ZnS NaCl CsCl NM (π)-afm FM AFMD (πππ)-afm FM (π)-afm E th (Ry) a th (Å) S Fe (Å) S N (Å) S E (Å) µ Fe (µ B ) N(E F )
12 Figure Caption Figure : Schematic illustration of FeN with NaCl (left), ZnS (middle) and CsCl (right) structures. Figure 2: Calculated total energy vs. volume for FeN with ZnS, NaCl and CsCl structures. Here (,, π)-afm and (π, π, π)-afm denote phases showing antiferromagnetic coupling along []- and []-directions, respectively. AFMD is the AFM state which consists of double layers with FM interlayer coupling along []-direction. The arrows label the volume corresponding to experimental lattice constants of ZnS- and NaCl-type FeN. Figure 3: Spin-resolved DOS calculated for FeN with (a) nonmagnetic ZnS, (b) (,, π)-afm NaCl and (c) FM CsCl structures at the theoretical equilibrium volume. Solid lines represent the majority-spin DOS and dash lines the minority-spin DOS. The Fermi-level is at zero energy. Figure 4: Orbital-projected partial DOS calculated for FeN with (a) NM ZnS, (b) (,, π)-afm NaCl and (c) FM CsCl structures at theoretical equilibrium volume. The dash, dot, dash-dotted and solid lines represent the DOS projected for Fe s,p,d and N p orbitals. The Fermi-level is at zero energy. Figure 5: The COHP curves calculated for s-p, p-p and d-p interactions between Fe and N atoms in FeN with (a) NM ZnS, (b) and (c) (,,π)-afm NaCl and (d) FM CsCl structures. Here (b) and (c) show the Fe-N interactions within and between the sublattice in (,,π)-afm NaCl structure, respectively. The solid, dash-dotted and dash lines represent s-p, p-p and d-p interactions. The integrated COHP (in ev/cell) for d-p interaction are also plotted with thick solid lines. Figure 6: Calculated Fe magnetic moment in FeN with ZnS, NaCl and CsCl structures as function of the nearest-neighboring Fe-N distance. The arrows indicate the distances corresponding to experimental lattice constants of NaCl- and ZnS-type FeN. 2


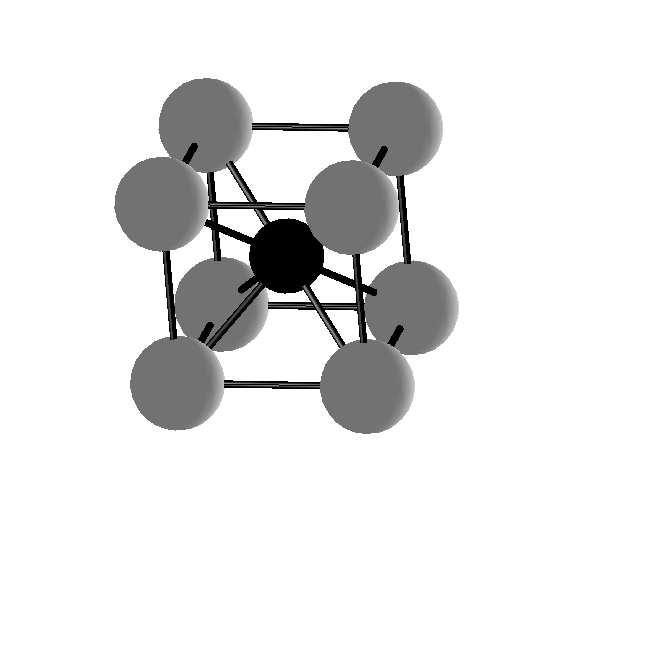
13 Fig. of Y. Kong 3
14 E tot (Ry / FeN) ZnS, NM ZnS, FM ZnS, π-afm CsCl, FM CsCl, π-afm V ZnS,expṾNaCl,exp. NaCl, NM NaCl, FM NaCl, π-afm NaCl, AFMD NaCl, πππ-afm Volume (a.u. 3 / FeN) Fig. 2 of Y. Kong 4
15 3.5 3 (a) (b) 4 DOS (states/ev spin FeN) (c) ENERGY (ev) Fig. 3 of Y. Kong 5
16 6 (a) (b) 4 DOS (states/ev FeN) (c) ENERGY (ev) Fig. 4 of Y. Kong 6
17 3 2.5 (a).2 (b) -COHP (/cell), -ICOHP (ev/cell) COHP (/cell), -ICOHP (ev/cell) ENERGY (ev) ENERGY (ev).6 (d).2 (c) -COHP (/cell), -ICOHP (ev/cell).4.2 -COHP (/cell), -ICOHP (ev/cell) ENERGY (ev) ENERGY (ev) Fig. 5 of Y. Kong 7
18 µ Fe ( µ B ) NaCl, FM NaCl, π-afm NaCl, AFMD NaCl, πππ-afm CsCl, FM CsCl, π-afm ZnS, FM ZnS, π-afm. d ZnS, exp d NaCl, exp d Fe-N (a.u.) Fig. 6 of Y. Kong 8
CHAPTER 6. ELECTRONIC AND MAGNETIC STRUCTURE OF ZINC-BLENDE TYPE CaX (X = P, As and Sb) COMPOUNDS
 COMPOUNDS") 143 CHAPTER 6 ELECTRONIC AND MAGNETIC STRUCTURE OF ZINC-BLENDE TYPE CaX (X = P, As and Sb) COMPOUNDS 6.1 INTRODUCTION Almost the complete search for possible magnetic materials has been performed utilizing
143 CHAPTER 6 ELECTRONIC AND MAGNETIC STRUCTURE OF ZINC-BLENDE TYPE CaX (X = P, As and Sb) COMPOUNDS 6.1 INTRODUCTION Almost the complete search for possible magnetic materials has been performed utilizing
Electronic structure of U 5 Ge 4
 Materials Science-Poland, Vol. 25, No. 2, 2007 Electronic structure of U 5 Ge 4 A. SZAJEK * Institute of Molecular Physics, Polish Academy of Sciences, ul. Smoluchowskiego 17, 60-179 Poznań, Poland U 5
Materials Science-Poland, Vol. 25, No. 2, 2007 Electronic structure of U 5 Ge 4 A. SZAJEK * Institute of Molecular Physics, Polish Academy of Sciences, ul. Smoluchowskiego 17, 60-179 Poznań, Poland U 5
Self-compensating incorporation of Mn in Ga 1 x Mn x As
 Self-compensating incorporation of Mn in Ga 1 x Mn x As arxiv:cond-mat/0201131v1 [cond-mat.mtrl-sci] 9 Jan 2002 J. Mašek and F. Máca Institute of Physics, Academy of Sciences of the CR CZ-182 21 Praha
Self-compensating incorporation of Mn in Ga 1 x Mn x As arxiv:cond-mat/0201131v1 [cond-mat.mtrl-sci] 9 Jan 2002 J. Mašek and F. Máca Institute of Physics, Academy of Sciences of the CR CZ-182 21 Praha
First Principles Calculation of Defect and Magnetic Structures in FeCo
 Materials Transactions, Vol. 47, No. 11 (26) pp. 2646 to 26 Special Issue on Advances in Computational Materials Science and Engineering IV #26 The Japan Institute of Metals First Principles Calculation
Materials Transactions, Vol. 47, No. 11 (26) pp. 2646 to 26 Special Issue on Advances in Computational Materials Science and Engineering IV #26 The Japan Institute of Metals First Principles Calculation
FULL POTENTIAL LINEARIZED AUGMENTED PLANE WAVE (FP-LAPW) IN THE FRAMEWORK OF DENSITY FUNCTIONAL THEORY
 IN THE FRAMEWORK OF DENSITY FUNCTIONAL THEORY") FULL POTENTIAL LINEARIZED AUGMENTED PLANE WAVE (FP-LAPW) IN THE FRAMEWORK OF DENSITY FUNCTIONAL THEORY C.A. Madu and B.N Onwuagba Department of Physics, Federal University of Technology Owerri, Nigeria
FULL POTENTIAL LINEARIZED AUGMENTED PLANE WAVE (FP-LAPW) IN THE FRAMEWORK OF DENSITY FUNCTIONAL THEORY C.A. Madu and B.N Onwuagba Department of Physics, Federal University of Technology Owerri, Nigeria
CHAPTER 4. ELECTRONIC AND MAGNETIC PROPERTIES OF MX 2 (M = V, Nb; X = Al, Ga, In, Cl, Br AND I) COMPOUNDS IN CdI 2 -TYPE STRUCTURE
 COMPOUNDS IN CdI 2 -TYPE STRUCTURE") 84 CHAPTER 4 ELECTRONIC AND MAGNETIC PROPERTIES OF MX 2 (M = V, Nb; X = Al, Ga, In, Cl, Br AND I) COMPOUNDS IN CdI 2 -TYPE STRUCTURE 4.1 INTRODUCTION As ideal materials for use in spintronic devices, the
84 CHAPTER 4 ELECTRONIC AND MAGNETIC PROPERTIES OF MX 2 (M = V, Nb; X = Al, Ga, In, Cl, Br AND I) COMPOUNDS IN CdI 2 -TYPE STRUCTURE 4.1 INTRODUCTION As ideal materials for use in spintronic devices, the
Electronic and Bonding Properties of Half-metallic PtMnSb and NiMnSb : First Principles Study
 J. Pure Appl. & Ind. Phys. Vol.2 (3), 278-285 (2012) Electronic and Bonding Properties of Half-metallic PtMnSb and NiMnSb : First Principles Study I. B. SHAMEEM BANU Department of Physics, B.S. Abdur Rahman
J. Pure Appl. & Ind. Phys. Vol.2 (3), 278-285 (2012) Electronic and Bonding Properties of Half-metallic PtMnSb and NiMnSb : First Principles Study I. B. SHAMEEM BANU Department of Physics, B.S. Abdur Rahman
ELECTRONIC AND MAGNETIC PROPERTIES OF BERKELIUM MONONITRIDE BKN: A FIRST- PRINCIPLES STUDY
 ELECTRONIC AND MAGNETIC PROPERTIES OF BERKELIUM MONONITRIDE BKN: A FIRST- PRINCIPLES STUDY Gitanjali Pagare Department of Physics, Sarojini Naidu Govt. Girls P. G. Auto. College, Bhopal ( India) ABSTRACT
ELECTRONIC AND MAGNETIC PROPERTIES OF BERKELIUM MONONITRIDE BKN: A FIRST- PRINCIPLES STUDY Gitanjali Pagare Department of Physics, Sarojini Naidu Govt. Girls P. G. Auto. College, Bhopal ( India) ABSTRACT
Ferromagnetism in Cr-Doped GaN: A First Principles Calculation
 Ferromagnetism in Cr-Doped GaN: A First Principles Calculation G. P. Das, 1,2 B. K. Rao, 1 and P. Jena 1 1 Department of Physics, Virginia Commonwealth University Richmond, VA 23284-2000 2 Bhabha Atomic
Ferromagnetism in Cr-Doped GaN: A First Principles Calculation G. P. Das, 1,2 B. K. Rao, 1 and P. Jena 1 1 Department of Physics, Virginia Commonwealth University Richmond, VA 23284-2000 2 Bhabha Atomic
* Theoretische Physik II, Universitat Dortmund, Dortmund, Germany
 JOURNAL DE PHYSIQUE IV Colloque C8, suppl6ment au Journal de Physique III, Volume 5, dkembre 1995 Structural Phase Transformation and Phonon Softening in Iron-Based Alloys H.C. Herper, E. Hoffmann, P.
JOURNAL DE PHYSIQUE IV Colloque C8, suppl6ment au Journal de Physique III, Volume 5, dkembre 1995 Structural Phase Transformation and Phonon Softening in Iron-Based Alloys H.C. Herper, E. Hoffmann, P.
Bonding and Elastic Properties in Ti 2 AC (A = Ga or Tl)
") Printed in the Republic of Korea http://dx.doi.org/10.5012/jkcs.2013.57.1.35 Bonding and Elastic Properties in Ti 2 AC (A = Ga or Tl) Dae-Bok Kang* Department of Chemistry, Kyungsung University, Busan
Printed in the Republic of Korea http://dx.doi.org/10.5012/jkcs.2013.57.1.35 Bonding and Elastic Properties in Ti 2 AC (A = Ga or Tl) Dae-Bok Kang* Department of Chemistry, Kyungsung University, Busan
Moldavian Journal of the Physical Sciences, N2, 2002
 Moldavian Journal of the Physical Sciences, N2, 2 LCTRONIC STRUCTURS AND MAGNTIC PROPRTIS O R n+1 Co n+ 2n (n=, 1, 2, and ) COMPOUNDS WITH R=Y AND P. Vlaic,. urzo aculty of Physics, abes-olyai University,
Moldavian Journal of the Physical Sciences, N2, 2 LCTRONIC STRUCTURS AND MAGNTIC PROPRTIS O R n+1 Co n+ 2n (n=, 1, 2, and ) COMPOUNDS WITH R=Y AND P. Vlaic,. urzo aculty of Physics, abes-olyai University,
The Augmented Spherical Wave Method
 Introduction Institut für Physik, Universität Augsburg Electronic Structure in a Nutshell Outline 1 Fundamentals Generations 2 Outline 1 Fundamentals Generations 2 Outline Fundamentals Generations 1 Fundamentals
Introduction Institut für Physik, Universität Augsburg Electronic Structure in a Nutshell Outline 1 Fundamentals Generations 2 Outline 1 Fundamentals Generations 2 Outline Fundamentals Generations 1 Fundamentals
Ceramic Bonding. CaF 2 : large SiC: small
 Recall ceramic bonding: - Mixed ionic and covalent. - % ionic character ( f ) increases with difference in electronegativity Large vs small ionic bond character: Ceramic Bonding CaF : large SiC: small
Recall ceramic bonding: - Mixed ionic and covalent. - % ionic character ( f ) increases with difference in electronegativity Large vs small ionic bond character: Ceramic Bonding CaF : large SiC: small
Electronic structure, magnetic structure, and metalatom site preferences in CrMnAs
 Graduate Theses and Dissertations Graduate College 2013 Electronic structure, magnetic structure, and metalatom site preferences in CrMnAs Laura Christine Lutz Iowa State University Follow this and additional
Graduate Theses and Dissertations Graduate College 2013 Electronic structure, magnetic structure, and metalatom site preferences in CrMnAs Laura Christine Lutz Iowa State University Follow this and additional
arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 13 Nov 2003
![arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 13 Nov 2003](/thumbs/88/115260649.jpg "arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 13 Nov 2003") 1. 14 August 1996 (final accepted version arxiv:cond-mat/0311297v1 [cond-mat.mtrl-sci] 13 Nov 2003 2. Non-collinear magnetism in distorted perovskite compounds 3. I.V.Solovyev a,, N.Hamada b, K.Terakura
1. 14 August 1996 (final accepted version arxiv:cond-mat/0311297v1 [cond-mat.mtrl-sci] 13 Nov 2003 2. Non-collinear magnetism in distorted perovskite compounds 3. I.V.Solovyev a,, N.Hamada b, K.Terakura
PBS: FROM SOLIDS TO CLUSTERS
 PBS: FROM SOLIDS TO CLUSTERS E. HOFFMANN AND P. ENTEL Theoretische Tieftemperaturphysik Gerhard-Mercator-Universität Duisburg, Lotharstraße 1 47048 Duisburg, Germany Semiconducting nanocrystallites like
PBS: FROM SOLIDS TO CLUSTERS E. HOFFMANN AND P. ENTEL Theoretische Tieftemperaturphysik Gerhard-Mercator-Universität Duisburg, Lotharstraße 1 47048 Duisburg, Germany Semiconducting nanocrystallites like
Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC
 286 Brazilian Journal of Physics, vol. 36, no. 2A, June, 2006 Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC Clas Persson and Susanne Mirbt Department
286 Brazilian Journal of Physics, vol. 36, no. 2A, June, 2006 Improved Electronic Structure and Optical Properties of sp-hybridized Semiconductors Using LDA+U SIC Clas Persson and Susanne Mirbt Department
First-principle Study for Al x Ga 1-x P and Mn-doped AlGaP 2 Electronic Properties
 Journal of Magnetics 20(4), 331-335 (2015) ISSN (Print) 1226-1750 ISSN (Online) 2233-6656 http://dx.doi.org/10.4283/jmag.2015.20.4.331 First-principle Study for Al x Ga 1-x P and Mn-doped AlGaP 2 Electronic
Journal of Magnetics 20(4), 331-335 (2015) ISSN (Print) 1226-1750 ISSN (Online) 2233-6656 http://dx.doi.org/10.4283/jmag.2015.20.4.331 First-principle Study for Al x Ga 1-x P and Mn-doped AlGaP 2 Electronic
Department of Physics, Anna University, Sardar Patel Road, Guindy, Chennai -25, India.
 Advanced Materials Research Online: 2013-02-13 ISSN: 1662-8985, Vol. 665, pp 43-48 doi:10.4028/www.scientific.net/amr.665.43 2013 Trans Tech Publications, Switzerland Electronic Structure and Ground State
Advanced Materials Research Online: 2013-02-13 ISSN: 1662-8985, Vol. 665, pp 43-48 doi:10.4028/www.scientific.net/amr.665.43 2013 Trans Tech Publications, Switzerland Electronic Structure and Ground State
N. Gonzalez Szwacki and Jacek A. Majewski Faculty of Physics, University of Warsaw, ul. Hoża 69, Warszawa, Poland
 Ab initio studies of Co 2 FeAl 1-x Si x Heusler alloys N. Gonzalez Szwacki and Jacek A. Majewski Faculty of Physics, University of Warsaw, ul. Hoża 69, 00-681 Warszawa, Poland Abstract We present results
Ab initio studies of Co 2 FeAl 1-x Si x Heusler alloys N. Gonzalez Szwacki and Jacek A. Majewski Faculty of Physics, University of Warsaw, ul. Hoża 69, 00-681 Warszawa, Poland Abstract We present results
Experiment 7: Understanding Crystal Structures
 Experiment 7: Understanding Crystal Structures To do well in this laboratory experiment you need to be familiar with the concepts of lattice, crystal structure, unit cell, coordination number, the different
Experiment 7: Understanding Crystal Structures To do well in this laboratory experiment you need to be familiar with the concepts of lattice, crystal structure, unit cell, coordination number, the different
One-dimensional magnetism of one-dimensional metallic chains in bulk MnB 4.
 One-dimensional magnetism of one-dimensional metallic chains in bulk MnB 4. S. Khmelevskyi 1*, J. Redinger 1, A.B. Shick 2, and P. Mohn 1. 1 Institute of Applied Physics, CMS, Vienna University of Technology,
One-dimensional magnetism of one-dimensional metallic chains in bulk MnB 4. S. Khmelevskyi 1*, J. Redinger 1, A.B. Shick 2, and P. Mohn 1. 1 Institute of Applied Physics, CMS, Vienna University of Technology,
WORLD JOURNAL OF ENGINEERING
 Magnetism and half-metallicity of some Cr-based alloys and their potential for application in spintronic devices by Yong Liu, S. K. Bose and J. Kudrnovsk y reprinted from WORLD JOURNAL OF ENGINEERING VOLUME
Magnetism and half-metallicity of some Cr-based alloys and their potential for application in spintronic devices by Yong Liu, S. K. Bose and J. Kudrnovsk y reprinted from WORLD JOURNAL OF ENGINEERING VOLUME
NOT FOR DISTRIBUTION REVIEW COPY. Na 2 IrO 3 as a molecular orbital crystal
 Na 2 IrO 3 as a molecular orbital crystal I. I. Mazin, 1 Harald O. Jeschke, 2 Kateryna Foyevtsova, 2 Roser Valentí, 2 and D. I. Khomskii 3 1 Code 6393, Naval Research Laboratory, Washington, DC 237, USA
Na 2 IrO 3 as a molecular orbital crystal I. I. Mazin, 1 Harald O. Jeschke, 2 Kateryna Foyevtsova, 2 Roser Valentí, 2 and D. I. Khomskii 3 1 Code 6393, Naval Research Laboratory, Washington, DC 237, USA
CHAPTER: 8. ELECTRONIC STRUCTURE AND ELASTIC PROPERTIES OF CrC AND CrN. 8.1 Introduction. Ph.D. Thesis: J. Maibam
 CHAPTER -8 CHAPTER: 8 ELECTRONIC STRUCTURE AND ELASTIC PROPERTIES OF CrC AND CrN 8.1 Introduction In this chapter, we have selected CrC and CrN from group VIB transition metal carbides and nitrides for
CHAPTER -8 CHAPTER: 8 ELECTRONIC STRUCTURE AND ELASTIC PROPERTIES OF CrC AND CrN 8.1 Introduction In this chapter, we have selected CrC and CrN from group VIB transition metal carbides and nitrides for
Electronic structure of barium titanate : an abinitio DFT study. Hong-Jian Feng, Fa-Min Liu
 Electronic structure of barium titanate : an abinitio DFT study Hong-Jian Feng, Fa-Min Liu Department of Physics, School of Sciences, Beijing University of Aeronautics & Astronautics, Beijing 183, P. R.
Electronic structure of barium titanate : an abinitio DFT study Hong-Jian Feng, Fa-Min Liu Department of Physics, School of Sciences, Beijing University of Aeronautics & Astronautics, Beijing 183, P. R.
arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 7 Feb 2002
![arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 7 Feb 2002](/thumbs/92/108939501.jpg "arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 7 Feb 2002") Electronic structures of doped anatase TiO : Ti x M x O (M=Co, Mn, Fe, Ni) Min Sik Park, S. K. Kwon, and B. I. Min Department of Physics and electron Spin Science Center, Pohang University of Science and
Electronic structures of doped anatase TiO : Ti x M x O (M=Co, Mn, Fe, Ni) Min Sik Park, S. K. Kwon, and B. I. Min Department of Physics and electron Spin Science Center, Pohang University of Science and
Structure and Curie temperature of Y 2 Fe 17 x Cr x
 Vol. 46 No. 4 SCIENCE IN CHINA (Series G) August 2003 Structure and Curie temperature of Y 2 Fe 17 x Cr x HAO Shiqiang ( ) 1 & CHEN Nanxian ( ) 1,2 1. Department of Physics, Tsinghua University, Beijing
Vol. 46 No. 4 SCIENCE IN CHINA (Series G) August 2003 Structure and Curie temperature of Y 2 Fe 17 x Cr x HAO Shiqiang ( ) 1 & CHEN Nanxian ( ) 1,2 1. Department of Physics, Tsinghua University, Beijing
DFT EXERCISES. FELIPE CERVANTES SODI January 2006
 DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
Chapter 6 Antiferromagnetism and Other Magnetic Ordeer
 Chapter 6 Antiferromagnetism and Other Magnetic Ordeer 6.1 Mean Field Theory of Antiferromagnetism 6.2 Ferrimagnets 6.3 Frustration 6.4 Amorphous Magnets 6.5 Spin Glasses 6.6 Magnetic Model Compounds TCD
Chapter 6 Antiferromagnetism and Other Magnetic Ordeer 6.1 Mean Field Theory of Antiferromagnetism 6.2 Ferrimagnets 6.3 Frustration 6.4 Amorphous Magnets 6.5 Spin Glasses 6.6 Magnetic Model Compounds TCD
The Gutzwiller Density Functional Theory
 The Gutzwiller Density Functional Theory Jörg Bünemann, BTU Cottbus I) Introduction 1. Model for an H 2 -molecule 2. Transition metals and their compounds II) Gutzwiller variational theory 1. Gutzwiller
The Gutzwiller Density Functional Theory Jörg Bünemann, BTU Cottbus I) Introduction 1. Model for an H 2 -molecule 2. Transition metals and their compounds II) Gutzwiller variational theory 1. Gutzwiller
Defects in TiO 2 Crystals
 , March 13-15, 2013, Hong Kong Defects in TiO 2 Crystals Richard Rivera, Arvids Stashans 1 Abstract-TiO 2 crystals, anatase and rutile, have been studied using Density Functional Theory (DFT) and the Generalized
, March 13-15, 2013, Hong Kong Defects in TiO 2 Crystals Richard Rivera, Arvids Stashans 1 Abstract-TiO 2 crystals, anatase and rutile, have been studied using Density Functional Theory (DFT) and the Generalized
Band calculations: Theory and Applications
 Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
Band calculations: Theory and Applications Lecture 2: Different approximations for the exchange-correlation correlation functional in DFT Local density approximation () Generalized gradient approximation
Electronic Structure and Magnetic Properties of Cu[C(CN) 3 ] 2 and Mn[C(CN) 3 ] 2 Based on First Principles
![Electronic Structure and Magnetic Properties of Cu[C(CN) 3 ] 2 and Mn[C(CN) 3 ] 2 Based on First Principles](/thumbs/75/71927831.jpg "Electronic Structure and Magnetic Properties of Cu[C(CN) 3 ] 2 and Mn[C(CN) 3 ] 2 Based on First Principles") Commun. Theor. Phys. (Beijing, China) 54 (2010) pp. 938 942 c Chinese Physical Society and IOP Publishing Ltd Vol. 54, No. 5, November 15, 2010 Electronic Structure and Magnetic Properties of Cu[C(CN)
Commun. Theor. Phys. (Beijing, China) 54 (2010) pp. 938 942 c Chinese Physical Society and IOP Publishing Ltd Vol. 54, No. 5, November 15, 2010 Electronic Structure and Magnetic Properties of Cu[C(CN)
CHAPTER 2. ELECTRONIC STRUCTURE AND GROUND STATE PROPERTIES OF M 2 O (M: Li, Na, K, Rb)
") 30 CHAPTER 2 ELECTRONIC STRUCTURE AND GROUND STATE PROPERTIES OF M 2 O (M: Li, Na, K, Rb) 2.1 INTRODUCTION Oxides of alkali metals (M 2 O) (M: Li, Na, K, Rb) play an important role in reducing the work
30 CHAPTER 2 ELECTRONIC STRUCTURE AND GROUND STATE PROPERTIES OF M 2 O (M: Li, Na, K, Rb) 2.1 INTRODUCTION Oxides of alkali metals (M 2 O) (M: Li, Na, K, Rb) play an important role in reducing the work
Theoretical study of superconductivity in MgB 2 and its alloys
 Bull. Mater. Sci., Vol. 26, No. 1, January 2003, pp. 131 135. Indian Academy of Sciences. Theoretical study of superconductivity in MgB 2 and its alloys P P SINGH Department of Physics, Indian Institute
Bull. Mater. Sci., Vol. 26, No. 1, January 2003, pp. 131 135. Indian Academy of Sciences. Theoretical study of superconductivity in MgB 2 and its alloys P P SINGH Department of Physics, Indian Institute
Introduction to Solid State Physics or the study of physical properties of matter in a solid phase
 Introduction to Solid State Physics or the study of physical properties of matter in a solid phase Prof. Germar Hoffmann 1. Crystal Structures 2. Reciprocal Lattice 3. Crystal Binding and Elastic Constants
Introduction to Solid State Physics or the study of physical properties of matter in a solid phase Prof. Germar Hoffmann 1. Crystal Structures 2. Reciprocal Lattice 3. Crystal Binding and Elastic Constants
'~IMI 1225 H 11A - LA Q I UIII 1111=11.61 MICROCOPY RESOLUTION TEST CHART NATIONAL BUREAU OF STANDARDS A ...
 RD-A15i 269 THEORY OF MAGNETIC AND STRUCTURAL ORDERING IN IRONU / MARYLAND UNIV COLLEGE PARK DEPT OF PHYSICS AND ASTRONOMY C S WANG ET AL. 1984 NSF-DMR82-13768 UNCLASSIFIED F/G 11/6 NL!'EmN mhhhh '~IMI
RD-A15i 269 THEORY OF MAGNETIC AND STRUCTURAL ORDERING IN IRONU / MARYLAND UNIV COLLEGE PARK DEPT OF PHYSICS AND ASTRONOMY C S WANG ET AL. 1984 NSF-DMR82-13768 UNCLASSIFIED F/G 11/6 NL!'EmN mhhhh '~IMI
Quantum Chemical Study of Defective Chromium Oxide
 , March 13-15, 2013, Hong Kong Quantum Chemical Study of Defective Chromium Oxide Richard Rivera, Soraya Jácome, Frank Maldonado, Arvids Stashans 1 Abstract Through the use of first-principles calculations
, March 13-15, 2013, Hong Kong Quantum Chemical Study of Defective Chromium Oxide Richard Rivera, Soraya Jácome, Frank Maldonado, Arvids Stashans 1 Abstract Through the use of first-principles calculations
All-Electron Full-Potential Calculations at O(ASA) Speed A Fata Morgana?
 Speed A Fata Morgana?") All-Electron Full-Potential Calculations at O(ASA) Speed A Fata Morgana? SFB 484, Teilprojekt D6 October 5, 2007 Outline 1 2 3 Outline 1 2 3 Outline 1 2 3 Outline 1 2 3 Back in the 1930 s... John C. Slater
All-Electron Full-Potential Calculations at O(ASA) Speed A Fata Morgana? SFB 484, Teilprojekt D6 October 5, 2007 Outline 1 2 3 Outline 1 2 3 Outline 1 2 3 Outline 1 2 3 Back in the 1930 s... John C. Slater
Basics of DFT applications to solids and surfaces
 Basics of DFT applications to solids and surfaces Peter Kratzer Physics Department, University Duisburg-Essen, Duisburg, Germany E-mail: Peter.Kratzer@uni-duisburg-essen.de Periodicity in real space and
Basics of DFT applications to solids and surfaces Peter Kratzer Physics Department, University Duisburg-Essen, Duisburg, Germany E-mail: Peter.Kratzer@uni-duisburg-essen.de Periodicity in real space and
Ab Initio Study of the 57 Fe Electric Field Gradient in (FeAl) 1 x T x (T = 3d Element) Dilute Alloys with B2-Type Structure
 1 x T x (T = 3d Element) Dilute Alloys with B2-Type Structure") Vol. 114 (2008) ACTA PHYSICA POLONICA A No. 6 Proceedings of the Polish Mössbauer Community Meeting 2008 Ab Initio Study of the 57 Fe Electric Field Gradient in (FeAl) 1 x T x (T = 3d Element) Dilute Alloys
Vol. 114 (2008) ACTA PHYSICA POLONICA A No. 6 Proceedings of the Polish Mössbauer Community Meeting 2008 Ab Initio Study of the 57 Fe Electric Field Gradient in (FeAl) 1 x T x (T = 3d Element) Dilute Alloys
Interstitial Mn in (Ga,Mn)As: Hybridization with Conduction Band and Electron Mediated Exchange Coupling
As: Hybridization with Conduction Band and Electron Mediated Exchange Coupling") Vol. 112 (2007) ACTA PHYSICA POLONICA A No. 2 Proceedings of the XXXVI International School of Semiconducting Compounds, Jaszowiec 2007 Interstitial Mn in (Ga,Mn)As: Hybridization with Conduction Band
Vol. 112 (2007) ACTA PHYSICA POLONICA A No. 2 Proceedings of the XXXVI International School of Semiconducting Compounds, Jaszowiec 2007 Interstitial Mn in (Ga,Mn)As: Hybridization with Conduction Band
Magnetism of close packed Fe 147 clusters
 Phase Transitions Vol. 00, No. 00, June 2005, 1 10 Magnetism of close packed Fe 147 clusters M. E. Gruner, G. Rollmann, S. Sahoo and P. Entel a Theoretical Physics, Universität Duisburg-Essen, Campus Duisburg,
Phase Transitions Vol. 00, No. 00, June 2005, 1 10 Magnetism of close packed Fe 147 clusters M. E. Gruner, G. Rollmann, S. Sahoo and P. Entel a Theoretical Physics, Universität Duisburg-Essen, Campus Duisburg,
Magnetic properties of spherical fcc clusters with radial surface anisotropy
 Magnetic properties of spherical fcc clusters with radial surface anisotropy D. A. Dimitrov and G. M. Wysin Department of Physics Kansas State University Manhattan, KS 66506-2601 (December 6, 1994) We
Magnetic properties of spherical fcc clusters with radial surface anisotropy D. A. Dimitrov and G. M. Wysin Department of Physics Kansas State University Manhattan, KS 66506-2601 (December 6, 1994) We
Electronic structure of Ce 2 Rh 3 Al 9
 Materials Science-Poland, Vol. 24, No. 3, 2006 Electronic structure of Ce 2 Rh 3 Al 9 J. GORAUS 1*, A. ŚLEBARSKI 1, J. DENISZCZYK 2 1 Institute of Physics, University of Silesia, ul. Bankowa 12, 40-007
Materials Science-Poland, Vol. 24, No. 3, 2006 Electronic structure of Ce 2 Rh 3 Al 9 J. GORAUS 1*, A. ŚLEBARSKI 1, J. DENISZCZYK 2 1 Institute of Physics, University of Silesia, ul. Bankowa 12, 40-007
Pressure dependence of Curie temperature and resistivity of complex Heusler alloys
 DPG Frühjahrstagung Berlin, 25. - 30. März 2012 Sektion Kondensierte Materie (SKM), 2012 p. 1 Pressure dependence of Curie temperature and resistivity of complex Heusler alloys Václav Drchal Institute
DPG Frühjahrstagung Berlin, 25. - 30. März 2012 Sektion Kondensierte Materie (SKM), 2012 p. 1 Pressure dependence of Curie temperature and resistivity of complex Heusler alloys Václav Drchal Institute
Supporting Information. Potential semiconducting and superconducting metastable Si 3 C. structures under pressure
 Supporting Information Potential semiconducting and superconducting metastable Si 3 C structures under pressure Guoying Gao 1,3,* Xiaowei Liang, 1 Neil W. Ashcroft 2 and Roald Hoffmann 3,* 1 State Key
Supporting Information Potential semiconducting and superconducting metastable Si 3 C structures under pressure Guoying Gao 1,3,* Xiaowei Liang, 1 Neil W. Ashcroft 2 and Roald Hoffmann 3,* 1 State Key
Orbital correlation and magnetocrystalline anisotropy in one-dimensional transition-metal systems
 PHYSICAL REVIEW B VOLUME 60, NUMBER 13 1 OCTOBER 1999-I Orbital correlation and magnetocrystalline anisotropy in one-dimensional transition-metal systems Lei Zhou Dingsheng Wang and Institute of Physics,
PHYSICAL REVIEW B VOLUME 60, NUMBER 13 1 OCTOBER 1999-I Orbital correlation and magnetocrystalline anisotropy in one-dimensional transition-metal systems Lei Zhou Dingsheng Wang and Institute of Physics,
status solidi Structural and electronic properties of ScSb, ScAs, ScP and ScN
 physica pss status solidi basic solid state physics b Structural and electronic properties of ScSb, ScAs, ScP and ScN AbdelGhani Tebboune 1, Djamel Rached, AbdelNour Benzair 3, Nadir Sekkal, 5,, and A.
physica pss status solidi basic solid state physics b Structural and electronic properties of ScSb, ScAs, ScP and ScN AbdelGhani Tebboune 1, Djamel Rached, AbdelNour Benzair 3, Nadir Sekkal, 5,, and A.
First-principles calculations for vacancy formation energies in Cu and Al; non-local e ect beyond the LSDA and lattice distortion
 Computational Materials Science 14 (1999) 56±61 First-principles calculations for vacancy formation energies in Cu and Al; non-local e ect beyond the LSDA and lattice distortion T. Hoshino a, *, N. Papanikolaou
Computational Materials Science 14 (1999) 56±61 First-principles calculations for vacancy formation energies in Cu and Al; non-local e ect beyond the LSDA and lattice distortion T. Hoshino a, *, N. Papanikolaou
Calculation and Analysis of the Dielectric Functions for BaTiO 3, PbTiO 3, and PbZrO 3
 CHINESE JOURNAL OF PHYSICS VOL. 1, NO. 3 June 213 Calculation and Analysis of the Dielectric Functions for BaTiO 3, PbTiO 3, and PbZrO 3 Chao Zhang and Dashu Yu School of Physics & Electronic Information
CHINESE JOURNAL OF PHYSICS VOL. 1, NO. 3 June 213 Calculation and Analysis of the Dielectric Functions for BaTiO 3, PbTiO 3, and PbZrO 3 Chao Zhang and Dashu Yu School of Physics & Electronic Information
Lattice Expansion of (Ga,Mn)As: The Role of Substitutional Mn and of the Compensating Defects
As: The Role of Substitutional Mn and of the Compensating Defects") Vol. 108 (2005) ACTA PHYSICA POLONICA A No. 5 Proceedings of the XXXIV International School of Semiconducting Compounds, Jaszowiec 2005 Lattice Expansion of (Ga,Mn)As: The Role of Substitutional Mn and
Vol. 108 (2005) ACTA PHYSICA POLONICA A No. 5 Proceedings of the XXXIV International School of Semiconducting Compounds, Jaszowiec 2005 Lattice Expansion of (Ga,Mn)As: The Role of Substitutional Mn and
arxiv:cond-mat/ v1 [cond-mat.mes-hall] 19 Dec 2006
![arxiv:cond-mat/ v1 [cond-mat.mes-hall] 19 Dec 2006](/thumbs/91/104898931.jpg "arxiv:cond-mat/ v1 [cond-mat.mes-hall] 19 Dec 2006") arxiv:cond-mat/678v [cond-mat.mes-hall] 9 Dec 6 Abstract Electronic structure of the Au/benzene-,-dithiol/Au transport interface: Effects of chemical bonding U. Schwingenschlögl, C. Schuster Institut für
arxiv:cond-mat/678v [cond-mat.mes-hall] 9 Dec 6 Abstract Electronic structure of the Au/benzene-,-dithiol/Au transport interface: Effects of chemical bonding U. Schwingenschlögl, C. Schuster Institut für
University of Bristol. 1 Naval Research Laboratory 2 II. Physikalisches Institut, Universität zu Köln
 Charge ordering as alternative to Jahn-Teller distortion In collaboration with Michelle Johannes 1, Daniel Khomskii 2 (theory) and Mohsen Abd-Elmeguid et al 2, Radu Coldea et al 3 (experiment) 1 Naval
Charge ordering as alternative to Jahn-Teller distortion In collaboration with Michelle Johannes 1, Daniel Khomskii 2 (theory) and Mohsen Abd-Elmeguid et al 2, Radu Coldea et al 3 (experiment) 1 Naval
Disturbing the dimers: electron- and hole-doping in the nonmagnetic intermetallic insulator FeGa 3
 Disturbing the dimers: electron- and hole-doping in the nonmagnetic intermetallic insulator FeGa 3 Antia S. Botana, Yundi Quan, Warren E. Pickett University of California-Davis, Davis, California, USA
Disturbing the dimers: electron- and hole-doping in the nonmagnetic intermetallic insulator FeGa 3 Antia S. Botana, Yundi Quan, Warren E. Pickett University of California-Davis, Davis, California, USA
Topological edge states in a high-temperature superconductor FeSe/SrTiO 3 (001) film
 film") Topological edge states in a high-temperature superconductor FeSe/SrTiO 3 (001) film Z. F. Wang 1,2,3+, Huimin Zhang 2,4+, Defa Liu 5, Chong Liu 2, Chenjia Tang 2, Canli Song 2, Yong Zhong 2, Junping Peng
Topological edge states in a high-temperature superconductor FeSe/SrTiO 3 (001) film Z. F. Wang 1,2,3+, Huimin Zhang 2,4+, Defa Liu 5, Chong Liu 2, Chenjia Tang 2, Canli Song 2, Yong Zhong 2, Junping Peng
Interatomic exchange coupling of BCC iron
 Interatomic exchange coupling of BCC iron Hai Wang, Pui-Wai Ma and C. H. Woo * Department of Electronic and Information Engineering, The Hong Kong Polytechnic University, Hong Kong SAR, China Abstract
Interatomic exchange coupling of BCC iron Hai Wang, Pui-Wai Ma and C. H. Woo * Department of Electronic and Information Engineering, The Hong Kong Polytechnic University, Hong Kong SAR, China Abstract
Electronic Structure of CsCl-Type Transition Metal Alloys
 2119 Progress of Theoretical Physics, Vol. 48, No. 6B, December 1972 Electronic Structure of CsCl-Type Transition Metal Alloys Jiro YAMASHTA and Setsuro ASANO nstitute for Solid State Physics, University
2119 Progress of Theoretical Physics, Vol. 48, No. 6B, December 1972 Electronic Structure of CsCl-Type Transition Metal Alloys Jiro YAMASHTA and Setsuro ASANO nstitute for Solid State Physics, University
Electronic properties and Fermi surface for new Fe-free layered pnictide-oxide superconductor BaTi 2 Bi 2 O from first principles
 Pis ma v ZhETF, vol. 97, iss. 4, pp.248 252 c 2013 February 25 Electronic properties and Fermi surface for new Fe-free layered pnictide-oxide superconductor BaTi 2 Bi 2 O from first principles D. V. Suetin,
Pis ma v ZhETF, vol. 97, iss. 4, pp.248 252 c 2013 February 25 Electronic properties and Fermi surface for new Fe-free layered pnictide-oxide superconductor BaTi 2 Bi 2 O from first principles D. V. Suetin,
arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 14 Jul 1997
![arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 14 Jul 1997](/thumbs/92/110189151.jpg "arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 14 Jul 1997") Inverse versus Normal NiAs Structures as High Pressure Phases of FeO and MnO arxiv:cond-mat/9707139v1 [cond-mat.mtrl-sci] 14 Jul 1997 Z. Fang, K. Terakura, H. Sawada, T. Miyazaki & I. Solovyev JRCAT, Angstrom
Inverse versus Normal NiAs Structures as High Pressure Phases of FeO and MnO arxiv:cond-mat/9707139v1 [cond-mat.mtrl-sci] 14 Jul 1997 Z. Fang, K. Terakura, H. Sawada, T. Miyazaki & I. Solovyev JRCAT, Angstrom
Magnetism of 3d, 4d, and 5d transition-metal impurities on Pd 001 and Pt 001 surfaces
 PHYSICAL REVIEW B VOLUME 53, NUMBER 4 15 JANUARY 1996-II Magnetism of 3d, 4d, and 5d transition-metal impurities on Pd 001 and Pt 001 surfaces V. S. Stepanyuk Max-Planck-Institut für Mikrostrukturphysik,
PHYSICAL REVIEW B VOLUME 53, NUMBER 4 15 JANUARY 1996-II Magnetism of 3d, 4d, and 5d transition-metal impurities on Pd 001 and Pt 001 surfaces V. S. Stepanyuk Max-Planck-Institut für Mikrostrukturphysik,
X-Ray and Mössbauer Spectra and Electronic Structure of ScFe 2 Si 2 Compound
 Journal of Materials Science and Engineering B 5 (1-2) (2015) 42-49 doi: 10.17265/2161-6221/2015.1-2.004 D DAVID PUBLISHING X-Ray and Mössbauer Spectra and Electronic Structure of ScFe 2 Si 2 Compound
Journal of Materials Science and Engineering B 5 (1-2) (2015) 42-49 doi: 10.17265/2161-6221/2015.1-2.004 D DAVID PUBLISHING X-Ray and Mössbauer Spectra and Electronic Structure of ScFe 2 Si 2 Compound
SnO 2 Physical and Chemical Properties due to the Impurity Doping
 , March 13-15, 2013, Hong Kong SnO 2 Physical and Chemical Properties due to the Impurity Doping Richard Rivera, Freddy Marcillo, Washington Chamba, Patricio Puchaicela, Arvids Stashans Abstract First-principles
, March 13-15, 2013, Hong Kong SnO 2 Physical and Chemical Properties due to the Impurity Doping Richard Rivera, Freddy Marcillo, Washington Chamba, Patricio Puchaicela, Arvids Stashans Abstract First-principles
Structure stability and magnetic properties of Os n B(n = 11 20) clusters
 clusters") Bull. Mater. Sci., Vol. 38, No. 2, April 2015, pp. 425 434. c Indian Academy of Sciences. Structure stability and magnetic properties of Os n B(n = 11 20) clusters XIU-RONG ZHANG 1,, MINLUO 2, FU-XING
Bull. Mater. Sci., Vol. 38, No. 2, April 2015, pp. 425 434. c Indian Academy of Sciences. Structure stability and magnetic properties of Os n B(n = 11 20) clusters XIU-RONG ZHANG 1,, MINLUO 2, FU-XING
Energy Stabilities, Magnetic Properties, and Electronic Structures of Diluted Magnetic Semiconductor Zn 1 x Mn x S(001) Thin Films
 Thin Films") CHINESE JOURNAL OF CHEMICAL PHYSICS VOLUME 24, NUMBER 1 FEBRUARY 27, 2011 ARTICLE Energy Stabilities, Magnetic Properties, and Electronic Structures of Diluted Magnetic Semiconductor Zn 1 x Mn x S(001)
CHINESE JOURNAL OF CHEMICAL PHYSICS VOLUME 24, NUMBER 1 FEBRUARY 27, 2011 ARTICLE Energy Stabilities, Magnetic Properties, and Electronic Structures of Diluted Magnetic Semiconductor Zn 1 x Mn x S(001)
Supplementary Figure 1 Two-dimensional map of the spin-orbit coupling correction to the scalar-relativistic DFT/LDA band gap. The calculations were
 Supplementary Figure 1 Two-dimensional map of the spin-orbit coupling correction to the scalar-relativistic DFT/LDA band gap. The calculations were performed for the Platonic model of PbI 3 -based perovskites
Supplementary Figure 1 Two-dimensional map of the spin-orbit coupling correction to the scalar-relativistic DFT/LDA band gap. The calculations were performed for the Platonic model of PbI 3 -based perovskites
Metallic and Ionic Structures and Bonding
 Metallic and Ionic Structures and Bonding Ionic compounds are formed between elements having an electronegativity difference of about 2.0 or greater. Simple ionic compounds are characterized by high melting
Metallic and Ionic Structures and Bonding Ionic compounds are formed between elements having an electronegativity difference of about 2.0 or greater. Simple ionic compounds are characterized by high melting
arxiv:cond-mat/ v1 7 Aug 1996
 EXCHANGE COUPLING AND MAGNETIZATION PROFILES OF BINARY AND TERNARY MAGNETIC MULTILAYERS F. Süss, U. Krey 1 Institut für Physik II der Universität, D-93040 Regensburg, Germany arxiv:cond-mat/9608037v1 7
EXCHANGE COUPLING AND MAGNETIZATION PROFILES OF BINARY AND TERNARY MAGNETIC MULTILAYERS F. Süss, U. Krey 1 Institut für Physik II der Universität, D-93040 Regensburg, Germany arxiv:cond-mat/9608037v1 7
2.1 Experimental and theoretical studies
 Chapter 2 NiO As stated before, the first-row transition-metal oxides are among the most interesting series of materials, exhibiting wide variations in physical properties related to electronic structure.
Chapter 2 NiO As stated before, the first-row transition-metal oxides are among the most interesting series of materials, exhibiting wide variations in physical properties related to electronic structure.
Electronic Effects in CO Chemisorption on Pt-Pb Intermetallic Surfaces: A Theoretical Study
 J. Phys. Chem. C 2007, 111, 17357-17369 17357 Electronic Effects in CO Chemisorption on Pt-Pb Intermetallic Surfaces: A Theoretical Study Chinmoy Ranjan, Roald Hoffmann,* Francis J. DiSalvo, and Héctor
J. Phys. Chem. C 2007, 111, 17357-17369 17357 Electronic Effects in CO Chemisorption on Pt-Pb Intermetallic Surfaces: A Theoretical Study Chinmoy Ranjan, Roald Hoffmann,* Francis J. DiSalvo, and Héctor
The Nature of Laves Phases: An Explorative Investigation of the Nb-Co System
 The Nature of Laves Phases: An Explorative Investigation of the Nb-Co System Daniel Grüner, Frank Stein 1, Martin Palm 1, Joachim Konrad 1, Tadahiro Yokosawa 2, Alim Ormeci, Walter Schnelle, Osamu Terasaki
The Nature of Laves Phases: An Explorative Investigation of the Nb-Co System Daniel Grüner, Frank Stein 1, Martin Palm 1, Joachim Konrad 1, Tadahiro Yokosawa 2, Alim Ormeci, Walter Schnelle, Osamu Terasaki
First-Principles Calculation of Point Defects in Uranium Dioxide
 Materials Transactions, Vol. 47, No. 11 (26) pp. 2651 to 2657 Special Issue on Advances in Computational Materials Science and Engineering IV #26 The Japan Institute of Metals First-Principles Calculation
Materials Transactions, Vol. 47, No. 11 (26) pp. 2651 to 2657 Special Issue on Advances in Computational Materials Science and Engineering IV #26 The Japan Institute of Metals First-Principles Calculation
Half-metallicity in Rhodium doped Chromium Phosphide: An ab-initio study
 Half-metallicity in Rhodium doped Chromium Phosphide: An ab-initio study B. Amutha 1,*, R. Velavan 1 1 Department of Physics, Bharath Institute of Higher Education and Research (BIHER), Bharath University,
Half-metallicity in Rhodium doped Chromium Phosphide: An ab-initio study B. Amutha 1,*, R. Velavan 1 1 Department of Physics, Bharath Institute of Higher Education and Research (BIHER), Bharath University,
Atomic Arrangement. Primer Materials For Science Teaching Spring
 Atomic Arrangement Primer Materials For Science Teaching Spring 2016 31.3.2015 Levels of atomic arrangements No order In gases, for example the atoms have no order, they are randomly distributed filling
Atomic Arrangement Primer Materials For Science Teaching Spring 2016 31.3.2015 Levels of atomic arrangements No order In gases, for example the atoms have no order, they are randomly distributed filling
EFFECTIVE MAGNETIC HAMILTONIANS: ab initio determination
 ICSM212, Istanbul, May 3, 212, Theoretical Magnetism I, 17:2 p. 1 EFFECTIVE MAGNETIC HAMILTONIANS: ab initio determination Václav Drchal Institute of Physics ASCR, Praha, Czech Republic in collaboration
ICSM212, Istanbul, May 3, 212, Theoretical Magnetism I, 17:2 p. 1 EFFECTIVE MAGNETIC HAMILTONIANS: ab initio determination Václav Drchal Institute of Physics ASCR, Praha, Czech Republic in collaboration
Density functional calculations on the charge-ordered and valence-mixed modification of YBaFe 2 O 5
 PHYSICAL REVIEW B 79, 53 009 Density functional calculations on the charge-ordered and valence-mixed modification of YBaFe O 5 Christian Spiel, Peter Blaha, and Karlheinz Schwarz Department of Materials
PHYSICAL REVIEW B 79, 53 009 Density functional calculations on the charge-ordered and valence-mixed modification of YBaFe O 5 Christian Spiel, Peter Blaha, and Karlheinz Schwarz Department of Materials
Compositional trends in Ni-Mn-Ga Heusler alloys: first-principles approach
 MATEC Web of Conferences 33, 05005 ( 2015) DOI: 10.1051/ matecconf/ 20153305005 C Owned by the authors, published by EDP Sciences, 2015 Compositional trends in Ni-Mn-Ga Heusler alloys: first-principles
MATEC Web of Conferences 33, 05005 ( 2015) DOI: 10.1051/ matecconf/ 20153305005 C Owned by the authors, published by EDP Sciences, 2015 Compositional trends in Ni-Mn-Ga Heusler alloys: first-principles
Southeast University, Nanjing, China 2 Department of Applied Physics, Aalto University,
 Supplementary Information to Solubility of Boron, Carbon and Nitrogen in Transition Metals: Getting Insight into Trends from First-Principles Calculations Xiaohui Hu, 1,2 Torbjörn Björkman 2,3, Harri Lipsanen
Supplementary Information to Solubility of Boron, Carbon and Nitrogen in Transition Metals: Getting Insight into Trends from First-Principles Calculations Xiaohui Hu, 1,2 Torbjörn Björkman 2,3, Harri Lipsanen
arxiv:cond-mat/ v1 9 Aug 2006
 Correlation-Driven Charge Order at a Mott Insulator - Band Insulator Digital Interface Rossitza Pentcheva 1 and Warren E. Pickett 2 1 Department of Earth and Environmental Sciences, University of Munich,
Correlation-Driven Charge Order at a Mott Insulator - Band Insulator Digital Interface Rossitza Pentcheva 1 and Warren E. Pickett 2 1 Department of Earth and Environmental Sciences, University of Munich,
Support Information. For. Theoretical study of water adsorption and dissociation on Ta 3 N 5 (100) surfaces
 surfaces") Support Information For Theoretical study of water adsorption and dissociation on Ta 3 N 5 (100) surfaces Submitted to Physical Chemistry Chemical Physics by Jiajia Wang a, Wenjun Luo a, Jianyong Feng
Support Information For Theoretical study of water adsorption and dissociation on Ta 3 N 5 (100) surfaces Submitted to Physical Chemistry Chemical Physics by Jiajia Wang a, Wenjun Luo a, Jianyong Feng
Atomic Arrangement. Primer in Materials Spring
 Atomic Arrangement Primer in Materials Spring 2017 30.4.2017 1 Levels of atomic arrangements No order In gases, for example the atoms have no order, they are randomly distributed filling the volume to
Atomic Arrangement Primer in Materials Spring 2017 30.4.2017 1 Levels of atomic arrangements No order In gases, for example the atoms have no order, they are randomly distributed filling the volume to
E12 UNDERSTANDING CRYSTAL STRUCTURES
 E1 UNDERSTANDING CRYSTAL STRUCTURES 1 Introduction In this experiment, the structures of many elements and compounds are rationalized using simple packing models. The pre-work revises and extends the material
E1 UNDERSTANDING CRYSTAL STRUCTURES 1 Introduction In this experiment, the structures of many elements and compounds are rationalized using simple packing models. The pre-work revises and extends the material
CHEMISTRY. The correlation between structure and properties helps in discovering new solid materials with desired properties
 CHEMISTRY 1 The correlation between structure and properties helps in discovering new solid materials with desired properties like high temperature superconductors, magnetic materials, biodegradable polymers
CHEMISTRY 1 The correlation between structure and properties helps in discovering new solid materials with desired properties like high temperature superconductors, magnetic materials, biodegradable polymers
LEAD-CHALCOGENIDES UNDER PRESSURE: AB-INITIO STUDY
 International Conference on Ceramics, Bikaner, India International Journal of Modern Physics: Conference Series Vol. 22 (2013) 612 618 World Scientific Publishing Company DOI: 10.1142/S201019451301074X
International Conference on Ceramics, Bikaner, India International Journal of Modern Physics: Conference Series Vol. 22 (2013) 612 618 World Scientific Publishing Company DOI: 10.1142/S201019451301074X
arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 13 Sep 2002
![arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 13 Sep 2002](/thumbs/75/71832444.jpg "arxiv:cond-mat/ v1 [cond-mat.mtrl-sci] 13 Sep 2002") Violation of the Minimum H-H Separation Rule for Metal Hydrides P. Ravindran, P. Vajeeston, R. Vidya, A. Kjekshus, and H. Fjellvåg Department of Chemistry, University of Oslo, Box 1033, Blindern, N-0315,
Violation of the Minimum H-H Separation Rule for Metal Hydrides P. Ravindran, P. Vajeeston, R. Vidya, A. Kjekshus, and H. Fjellvåg Department of Chemistry, University of Oslo, Box 1033, Blindern, N-0315,
Magnetic moment of iron in metallic environments
 PHYSICAL REVIEW B VOLUME 61, NUMBER 1 1 JANUARY 2000-I Magnetic moment of iron in metallic environments G. W. Fernando Department of Physics, U-46, University of Connecticut, Storrs, Connecticut 06269;
PHYSICAL REVIEW B VOLUME 61, NUMBER 1 1 JANUARY 2000-I Magnetic moment of iron in metallic environments G. W. Fernando Department of Physics, U-46, University of Connecticut, Storrs, Connecticut 06269;
Solids. properties & structure
 Solids properties & structure Determining Crystal Structure crystalline solids have a very regular geometric arrangement of their particles the arrangement of the particles and distances between them is
Solids properties & structure Determining Crystal Structure crystalline solids have a very regular geometric arrangement of their particles the arrangement of the particles and distances between them is
Effect of the Mn clustering in (GaMn)N on the magnetic transition temperature
N on the magnetic transition temperature") Effect of the Mn clustering in (GaMn)N on the magnetic transition temperature L. M. Sandratskii and P. Bruno Max-Planck Institut für Mikrostrukturphysik, D-06120 Halle, Germany S. Mirbt Department of Physics,
Effect of the Mn clustering in (GaMn)N on the magnetic transition temperature L. M. Sandratskii and P. Bruno Max-Planck Institut für Mikrostrukturphysik, D-06120 Halle, Germany S. Mirbt Department of Physics,
Mössbauer studies on FeSe and FeTe
 Mössbauer studies on FeSe and FeTe Yoshikazu Mizuguchi a,b,c, Takao Furubayashi a, Keita Deguchi a,b,c, Shunsuke Tsuda a,b, Takahide Yamaguchi a,b and Yoshihiko Takano a,b,c a Superconducting Materials
Mössbauer studies on FeSe and FeTe Yoshikazu Mizuguchi a,b,c, Takao Furubayashi a, Keita Deguchi a,b,c, Shunsuke Tsuda a,b, Takahide Yamaguchi a,b and Yoshihiko Takano a,b,c a Superconducting Materials
Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type
 Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type Zhigang Gu, a Lars Heinke, a,* Christof Wöll a, Tobias Neumann,
Supplemental Material: Experimental and Theoretical Investigations of the Electronic Band Structure of Metal-Organic Framework of HKUST-1 Type Zhigang Gu, a Lars Heinke, a,* Christof Wöll a, Tobias Neumann,
Energy bands in solids. Some pictures are taken from Ashcroft and Mermin from Kittel from Mizutani and from several sources on the web.
 Energy bands in solids Some pictures are taken from Ashcroft and Mermin from Kittel from Mizutani and from several sources on the web. we are starting to remind p E = = mv 1 2 = k mv = 2 2 k 2m 2 Some
Energy bands in solids Some pictures are taken from Ashcroft and Mermin from Kittel from Mizutani and from several sources on the web. we are starting to remind p E = = mv 1 2 = k mv = 2 2 k 2m 2 Some
2 B B D (E) Paramagnetic Susceptibility. m s probability. A) Bound Electrons in Atoms
 Paramagnetic Susceptibility. m s probability. A) Bound Electrons in Atoms") Paramagnetic Susceptibility A) Bound Electrons in Atoms m s probability B +½ p ½e x Curie Law: 1/T s=½ + B ½ p + ½e +x With increasing temperature T the alignment of the magnetic moments in a B field is
Paramagnetic Susceptibility A) Bound Electrons in Atoms m s probability B +½ p ½e x Curie Law: 1/T s=½ + B ½ p + ½e +x With increasing temperature T the alignment of the magnetic moments in a B field is
Ferromagnetism in Mn Doped GaN: From Clusters to Crystals
 Ferromagnetism in Mn Doped GaN: From Clusters to Crystals G. P. Das, B.K. Rao, and P. Jena Physics Department, Virginia Commonwealth University Richmond, VA 23284-2000 Abstract The magnetic coupling between
Ferromagnetism in Mn Doped GaN: From Clusters to Crystals G. P. Das, B.K. Rao, and P. Jena Physics Department, Virginia Commonwealth University Richmond, VA 23284-2000 Abstract The magnetic coupling between
Study Of Electronic And Linear Optical Properties Of Indium Pnictides (InX, X = P, As, Sb)
") International Journal of Physics and Applications. ISSN 0974-3103 Volume 7, Number 1 (2015), pp. 9-14 International Research Publication House http://www.irphouse.com Study Of Electronic And Linear Optical
International Journal of Physics and Applications. ISSN 0974-3103 Volume 7, Number 1 (2015), pp. 9-14 International Research Publication House http://www.irphouse.com Study Of Electronic And Linear Optical
Epitaxial Growth of Mn on Si(111)
") 105 Chapter 7 Epitaxial Growth of Mn on Si(111) 7.1 Introduction There are a few reports and experiments concerning the adsoption of Mn on Si(111), where film growth with and without a Bi surfactant layer
105 Chapter 7 Epitaxial Growth of Mn on Si(111) 7.1 Introduction There are a few reports and experiments concerning the adsoption of Mn on Si(111), where film growth with and without a Bi surfactant layer
Novel Magnetic Properties of Carbon Nanotubes. Abstract
 Novel Magnetic Properties of Carbon Nanotubes Jian Ping Lu Department of Physics and Astronomy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599 jpl@physics.unc.edu arxiv:cond-mat/94779v1
Novel Magnetic Properties of Carbon Nanotubes Jian Ping Lu Department of Physics and Astronomy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599 jpl@physics.unc.edu arxiv:cond-mat/94779v1
Heisenberg Hamiltonian description of multiple-sublattice itinerant-electron systems: General considerations and applications to NiMnSb and MnAs
 Heisenberg Hamiltonian description of multiple-sublattice itinerant-electron systems: General considerations and applications to and MnAs L. M. Sandratskii* Max-Planck Institut für Mikrostrukturphysik,
Heisenberg Hamiltonian description of multiple-sublattice itinerant-electron systems: General considerations and applications to and MnAs L. M. Sandratskii* Max-Planck Institut für Mikrostrukturphysik,
Theoretical Concepts of Spin-Orbit Splitting
 Chapter 9 Theoretical Concepts of Spin-Orbit Splitting 9.1 Free-electron model In order to understand the basic origin of spin-orbit coupling at the surface of a crystal, it is a natural starting point
Chapter 9 Theoretical Concepts of Spin-Orbit Splitting 9.1 Free-electron model In order to understand the basic origin of spin-orbit coupling at the surface of a crystal, it is a natural starting point