Motif Finding Algorithms. Sudarsan Padhy IIIT Bhubaneswar
|
|
|
- Quentin Murphy
- 6 years ago
- Views:
Transcription
1 Motif Finding Algorithms Sudarsan Padhy IIIT Bhubaneswar
2 Outline Gene Regulation Regulatory Motifs The Motif Finding Problem Brute Force Motif Finding Consensus and Pattern Branching: Greedy Motif Search PMS: Exhaustive Motif Search Comparison of Motif Finding Algorithms Presentation of Our Work on Motif Finding


3 Central Dogma: DNA -> RNA -> Protein DNA CCTGAGCCAACTATTGATGAA transcription RNA CCUGAGCCAACUAUUGAUGAA translation Protein PEPTIDE

4 Splice Sites Almost all donor splice sites exhibit GU, Almost all acceptor splice site exhibit AG. Not all GUs and AGs are used as splice site, exploit that exons have higher GC content or that certain motifs are located nearby

5 Transcription Start Sites (TSS)
6 Combinatorial Gene Regulation A microarray experiment showed that when gene X is knocked out, 20 other genes are not expressed How can one gene have such drastic effects?
7 Regulatory Proteins Gene X encodes regulatory protein, a.k.a. a transcription factor (TF) The 20 unexpressed genes rely on gene X s TF to induce transcription A single TF may regulate multiple genes
8 Regulatory Regions Every gene contains a regulatory region (RR) typically stretching bp upstream of the transcriptional start site Located within the RR are the Transcription Factor Binding Sites (TFBS), also known as motifs, specific for a given transcription factor TFs influence gene expression by binding to a specific location in the respective gene s regulatory region - TFBS
9 Transcription Factor Binding Sites A TFBS can be located anywhere within the Regulatory Region. TFBS may vary slightly across different regulatory regions since non-essential bases could mutate
10 MOTIF A nucleotide / amino acid sequence pattern having biological significance Occurs repeatedly,either in same molecule or in many molecules. Found in upstream intergenic region Functionally important regions of genome( gene) can be recognized by searching such patterns Used for locating binding sites, regulatory signals, to control gene expression Identifying potential drug target sites
11 Motifs and Transcriptional Start Sites ATCCCG TTCCGG ATCCCG gene gene gene ATGCCG gene ATGCCC gene
12 Identifying Motifs Genes are turned on or off by regulatory proteins These proteins bind to upstream regulatory regions of genes to either attract or block an RNA polymerase Regulatory protein (TF) binds to a short DNA sequence called a motif (TFBS) So finding the same motif in multiple genes regulatory regions suggests a regulatory relationship amongst those genes
13 Identifying Motifs: Complications We do not know the motif sequence We do not know where it is located relative to the genes start Motifs can differ slightly from one gene to the next How to discern it from random motifs?
14 The Motif Finding Problem Given a random sample of DNA sequences: cctgatagacgctatctggctatccacgtacgtaggtcctctgtgcgaatctatgcgtttccaaccat agtactggtgtacatttgatacgtacgtacaccggcaacctgaaacaaacgctcagaaccagaagtgc aaacgtacgtgcaccctctttcttcgtggctctggccaacgagggctgatgtataagacgaaaatttt agcctccgatgtaagtcatagctgtaactattacctgccacccctattacatcttacgtacgtataca Ctgttatacaacgcgtcatggcggggtatgcgttttggtcgtcgtacgctcgatcgttaacgtacgtc Find the pattern that is implanted in each of the individual sequences, namely, the motif
15 The Motif Finding Problem (cont d) Additional information: The hidden sequence is of length 8 The pattern is not exactly the same in each array because random point mutations may occur in the sequences
16 The Motif Finding Problem (cont d) The patterns revealed with no mutations: cctgatagacgctatctggctatccacgtacgtaggtcctctgtgcgaatctatgcgtttccaact agtactggtgtacatttgatacgtacgtacaccggcaacctgaaacaaacgctcagaaccagaagt gc aaacgtacgtgcaccctctttcttcgtggctctggccaacgagggctgatgtataagacgaaaatt tt agcctccgatgtaagtcatagctgtaactattacctgccacccctattacatcttacgtacgtata ca ctgttatacaacgcgtcatggcggggtatgcgttttggtcgtcgtacgctcgatcgttaacgtacg tc acgtacgt Consensus String
17 The Motif Finding Problem (cont d) The patterns with 2 point mutations: cctgatagacgctatctggctatccaggtacttaggtcctctgtgcgaatctatgcgtttccaacc at agtactggtgtacatttgatccatacgtacaccggcaacctgaaacaaacgctcagaaccagaagt gc aaacgttagtgcaccctctttcttcgtggctctggccaacgagggctgatgtataagacgaaaatt tt agcctccgatgtaagtcatagctgtaactattacctgccacccctattacatcttacgtccatata ca ctgttatacaacgcgtcatggcggggtatgcgttttggtcgtcgtacgctcgatcgttaccgtacg Gc
18 The Motif Finding Problem (cont d) The patterns with 2 point mutations: cctgatagacgctatctggctatccaggtacttaggtcctctgtgcgaatctatgcgtttccaacc at agtactggtgtacatttgatccatacgtacaccggcaacctgaaacaaacgctcagaaccagaagt gc aaacgttagtgcaccctctttcttcgtggctctggccaacgagggctgatgtataagacgaaaatt tt agcctccgatgtaagtcatagctgtaactattacctgccacccctattacatcttacgtccatata ca ctgttatacaacgcgtcatggcggggtatgcgttttggtcgtcgtacgctcgatcgttaccgtacg Gc Can we still find the motif, now that we have 2 mutations?
19 Defining Motifs To define a motif, lets say we know where the motif starts in the sequence The motif start positions in their sequences can be represented as s = (s 1,s 2,s 3,,s t )
20 Motifs: Profiles and Consensus a G g t a c T t t Alignment t t G C c A t a c g a c g t T A g a c g t C c A C c g t a c g 0 A Profile 0 C G T Consensus A C G T A C G T Line up the patterns by their start indexes s = (s 1, s 2,, s t ) Construct matrix profile with frequencies of each nucleotide in columns Consensus nucleotide in each position has the highest score in column
21 Consensus Think of consensus as an ancestor motif, from which mutated motifs emerged The distance between a real motif and the consensus sequence is generally less than that for two real motifs
22 Consensus (cont d)
23 Evaluating Motifs We have a guess about the consensus sequence, but how good is this consensus? Need to introduce a scoring function to compare different guesses and choose the best one.
24 Defining Some Terms t - number of sample DNA sequences n - length of each DNA sequence DNA - sample of DNA sequences (t x n array) l - length of the motif (l-mer) s i - starting position of an l-mer in sequence i s=(s 1, s 2, s t ) - array of motif s starting positions
25 Parameters l = 8 DNA cctgatagacgctatctggctatccaggtacttaggtcctctgtgcgaatctatgcgtttcca accat t=5 agtactggtgtacatttgatccatacgtacaccggcaacctgaaacaaacgctcagaaccaga agtgc aaacgttagtgcaccctctttcttcgtggctctggccaacgagggctgatgtataagacgaaa atttt agcctccgatgtaagtcatagctgtaactattacctgccacccctattacatcttacgtccat ataca n = 69 s s 1 = 26 s 2 = 21 s 3 = 3 s 4 = 56 s 5 = 60 ctgttatacaacgcgtcatggcggggtatgcgttttggtcgtcgtacgctcgatcgttaccgt acggc
26 Scoring Motifs l Given s = (s 1, s t ) and DNA: Score(s,DNA) = l max i= 1 k { A, T, C, G} count( k, i) a G g t a c T t C c A t a c g t a c g t T A g t a c g t C c A t C c g t a c g G A C G T Consensus a c g t a c g t Score =30
27 The Motif Finding Problem If starting positions s=(s 1, s 2, s t ) are given, finding consensus is easy even with mutations in the sequences because we can simply construct the profile to find the motif (consensus) But the starting positions s are usually not given. How can we find the best profile matrix?
28 The Motif Finding Problem: Formulation Goal: Given a set of DNA sequences, find a set of l-mers, one from each sequence, that maximizes the consensus score Input: A t x n matrix of DNA, and l, the length of the pattern to find Output: An array of t starting positions s = (s 1, s 2, s t ) maximizing Score(s,DNA)
29 Motif finding The motif finding problem manifests itself in two forms Known motif and unknown position: given a motif of length l with d mismatches, find its position in a genomic/protein sequence. Unknown motif and unknown position: given a set of sequences each of length m find a motif of length l with d mismatches. (l,d)-motif problem Introduced by Pavel Pevzner in 2001 The problem is to determine the motif M of length l, given n nucleotide sequences each of length m, and each containing a planted variant of M. More precisely, each such planted variant is a substring M that is with up to d point mutations. (d << l)
30 Expected number of motifs Assume that the sequences are i.i.d Probability that a given l-mer (l length motif) C occurs with up-to d mutations at a given position of a random sequence is Then the expected number of length l motifs that occur with up to d mutations at least once in each of the t random length n sequences is: Given t sequences of length n each, l the length of the motif, using a Brute force approach the complexity is l (n l + 1) t = O(l n t ). For t = 10, n = 1000, l = 10 { Fastest Computer-Jaguar-1759 tera Flops} computations /( 3600x24x365 x1759)>20 Million Years
31 The Motif Finding Problem: Brute Force Solution Compute the scores for each possible combination of starting positions s The best score will determine the best profile and the consensus pattern in DNA The goal is to maximize Score(s,DNA) by varying the starting positions s i, where: s i = [1,, n-l+1] i = [1,, t]
32 BruteForceMotifSearch 1. BruteForceMotifSearch(DNA, t, n, l) 2. bestscore 0 3. for each s=(s 1,s 2,..., s t ) from (1,1... 1) to (n-l+1,..., n-l+1) 4. if (Score(s,DNA) > bestscore) 5. bestscore score(s, DNA) 6. bestmotif (s 1,s 2,..., s t ) 7. return bestmotif
33 Running Time of BruteForceMotifSearch Varying (n - l + 1) positions in each of t sequences, we re looking at (n - l + 1) t sets of starting positions For each set of starting positions, the scoring function makes l operations, so complexity is l (n l + 1) t = O(l n t ) That means that for t = 8, n = 1000, l = 10 we must perform approximately computations it will take billions years
34 CONSENSUS: Greedy Motif Search Find two closest l-mers in sequences 1 and 2 and forms 2 x l alignment matrix with Score(s,2,DNA) At each of the following t-2 iterations CONSENSUS finds a best l-mer in sequence i from the perspective of the already constructed (i-1) x l alignment matrix for the first (i-1) sequences In other words, it finds an l-mer in sequence i maximizing Score(s,i,DNA) under the assumption that the first (i-1) l-mers have been already chosen CONSENSUS sacrifices optimal solution for speed: in fact the bulk of the time is actually spent locating the first
35 Some Motif Finding Programs CONSENSUS MULTIPROFILER Hertz, Stromo Keich, Pevzner (1989) (2002) MITRA GibbsDNA Eskin, Pevzner Lawrence et al (2002) (1993) Pattern Branching MEME Bailey, Elkan (1995) Price, Pevzner (2003) RandomProjections Buhler, Tompa (2002)
36 Planted Motif Challenge Input: n sequences of length m each. Output: Motif M, of length l Variants of interest have a hamming distance of d from M

37 How to proceed? Exhaustive search? Run time is high
38 Best Neighbor Branch from the seed strings Find best neighbor - highest score Don t consider branches where the upper bound is not as good as best score so far
39 Scoring PatternBranching use total distance score: For each sequence S i in the sample S = {S 1,..., S n }, let d(a, S i ) = min{d(a, P) P S i }. Then the total distance of A from the sample is d(a, S) = Si S d(a, S i ). For a pattern A, let D=Neighbor(A) be the set of patterns which differ from A in exactly 1 position. We define BestNeighbor(A) as the pattern B D=Neighbor(A) with lowest total distance d(b, S).
40 PatternBranching Algorithm

41 PatternBranching Performance PatternBranching is faster than other patternbased algorithms Motif Challenge Problem: sample of n = 20 sequences N = 600 nucleotides long implanted pattern of length l = 15 k = 4 mutations
42 PMS (Planted Motif Search) Generate all possible l-mers from out of the input sequence S i. Let C i be the collection of these l-mers. Example: AAGTCAGGAGT C i = 3-mers: AAG AGT GTC TCA CAG AGG GGA GAG AGT
43 All patterns at Hamming distance d = 1 AAG AGT GTC TCA CAG AGG GGA GAG AGT CAG CGT ATC ACA AAG CGG AGA AAG CGT GAG GGT CTC CCA GAG TGG CGA CAG GGT TAG TGT TTC GCA TAG GGG TGA TAG TGT ACG ACT GAC TAA CCG ACG GAA GCG ACT AGG ATT GCC TGA CGG ATG GCA GGG ATT ATG AAT GGC TTA CTG AAG GTA GTG AAT AAC AGA GTA TCC CAA AGA GGC GAA AGA AAA AGC GTG TCG CAC AGT GGG GAC AGC AAT AGG GTT TCT CAT AGC GGT GAT AGG
44 Sort the lists AAG AGT GTC TCA CAG AGG GGA GAG AGT AAA AAT ATC ACA AAG AAG AGA AAG AAT AAC ACT CTC CCA CAA ACG CGA CAG ACT AAT AGA GAC GCA CAC AGA GAA GAA AGA ACG AGC GCC TAA CAT AGC GCA GAC AGC AGG AGG GGC TCC CCG AGT GGC GAT AGG ATG ATT GTA TCG CGG ATG GGG GCG ATT CAG CGT GTG TCT CTG CGG GGT GGG CGT GAG GGT GTT TGA GAG GGG GTA GTG GGT TAG TGT TTC TTA TAG TGG TGA TAG TGT
45 Eliminate duplicates AAG AGT GTC TCA CAG AGG GGA GAG AGT AAA AAT ATC ACA AAG AAG AGA AAG AAT AAC ACT CTC CCA CAA ACG CGA CAG ACT AAT AGA GAC GCA CAC AGA GAA GAA AGA ACG AGC GCC TAA CAT AGC GCA GAC AGC AGG AGG GGC TCC CCG AGT GGC GAT AGG ATG ATT GTA TCG CGG ATG GGG GCG ATT CAG CGT GTG TCT CTG CGG GGT GGG CGT GAG GGT GTT TGA GAG GGG GTA GTG GGT TAG TGT TTC TTA TAG TGG TGA TAG TGT
46 Find motif common to all lists Follow this procedure for all sequences Find the motif common all L i (once duplicates have been eliminated) This is the planted motif
47 Motif finding The motif finding problem manifests itself in two forms Known motif and unknown position: given a motif of length l with d mismatches, find its position in a genomic/protein sequence. Unknown motif and unknown position: given a set of sequences each of length m find a motif of length l with d mismatches. (l,d)-motif problem Introduced by Pavel Pevzner in 2001 The problem is to determine the motif M of length l, given n nucleotide sequences each of length m, and each containing a planted variant of M. More precisely, each such planted variant is a substring M that is with up to d point mutations. (d << l)
48 PMS Running Time It takes time to Generate variants Sort lists Find and eliminate duplicates Running time of this algorithm: w is the word length of the computer
49 REFERENCES 1. N.C.Jones,P.A.Pevzner-An Introduction to Bioinformatics Algorithms- MIT press P.A.Pevzner-Computational Molecular Biology-An Algoritmic Approach-MIT press D.Gusfield- Algorithms on Strings, Trees and Sequences- Computer Science and Computational Biology-Cambrigde Univ. press-1997
50 Other Methods Existing approaches with limitations The motif finding problem has been studied extensively for many years in the literature using different approaches. Statistical [Workman and Stormo(2000); Thijs et al.(2001); Wang and Stormo(2003); Xing et al.(2004); Regnier and Denise(2004); Sinha et al. [2004]; Siddharthan et al. (2005); Shida (2006 ); Leung and Chin (2006); Carmack et al.(2007)] The limitations of existing statistical approach is that it ignores evolutionary relationship which can give some information about where a motif is most likely to be found between very similar genomes.
51 Other Methods(continued) Heuristic approach [Liu et al.(2002); Buhler and Tompa(2002); Blanchette and Tompa (2002); Liu et al.(2004); Hu et al.(2006); Mendes et al.(2006); Wei and Jensen (2006); Peng et al. (2006)] has polynomial complexity but does not guarantee optimality, where as Combinatorial (exhaustive) approach guarantees optimality at the cost of combinatorial complexity. Exhaustive [Marsan and Sagot (2000); Pevzner and Sze (2000); Sinha and Tompa(2000); Apostolico et al.(2000); Eskin and Pevzner (2001); Liu et al.(2002 ); Sinha (2003); Wang et al.(2005); Hon and Jain (2006); ] Approach guarantees optimality at the cost of combinatorial complexity. Despite many studies this problem is far from being satisfactorily solved.
52 OUR METHODS (A. mohapatra,p.m.misra,s.padhy) Motif finding using Information-Entropy with Kullback-Leibler- Divergence International Journal of Computer Science and Network Security, Vol. 9 No. 1 pp , 2009(ISSN: ) Motif Search in DNA Sequences Using Generalized Suffix Tree IEEE Computer Society,( ISBN: , DOI: /ICIT ) Motif Prediction using Weighted Degree String Kernels with Shift and Mismatch ACM (ICAC3-09), Pages 56-61, ACM ( ISBN: DOI: Prediction of Motifs with Unknown Length and Allowable Mismatches using Kernel Based Approach
53 Motif finding using Information-Entropy with Kullback-Leibler-Divergence International Journal of Computer Science and Network Security, Vol. 9 No. 1 pp , 2009(ISSN: )
54 Related Motif finding Approaches Information entropy, first introduced by Claude E.Shannon in his paper A Mathematical theory of Communications in 1948 describes information content of a signal or event. Models based on Positional Weight Matrix [Bucher,1990] [CONSENSUS, Gibbs sampling, EM etc.] PWM can be created by counting the number of times a base is observed at each position Limitations Existing algorithms based on PWM consider the most popular element in each column of the alignment matrix, and completely ignore the rest which are comparatively weak (probability of occurrence is less). Models based on Information Entropy [Schneider, 1990][LOGOS,2004] From information entropy point of view all sequences (strong or weak) can contain different amount of information about the biological signal in various forms and thus can be a potential candidate for replacing the PWM model
55 Contd. Let Σ be an alphabet of size N, for DNA Σ = {A, C, G, T}. A symbol is received from an alphabet of size N, Letters are sent with equal probability Thus the information entropy contained in a symbol from an alphabet of size N is log 2 N, under uniform probability distribution. Close parallels between the mathematical expressions for the thermodynamic entropy S, and the Shannon information theoretic entropy H. The measure of information (Shannon entropy) is mathematically, similar to that of disorder (Boltzmann entropy) log H( p = I = 2 Nn 1,..., pn) n N = pi log2 pi i = 1
56 Motifs: Profiles and Consensus Alignment a G g t a c T t C c A t a c g t a c g t T A g t a c g t C c A t C c g t a c g G A Profile C G T Consensus A C G T A C G T Line up the patterns by their start indexes s = (s 1, s 2,, s t ) Construct matrix profile with frequencies of each nucleotide in columns Consensus nucleotide in each position has the highest score in column
57 PWM TTGTGGC TTGTGAT GCCTGAC TTTTGAT ATTTATT TTGTGAT AAGTGTC ATTTGCA ACGTGAT ATGTGAG TTGTGAT GTGTGAA SEQUENCES CTGTGAG ATGCAAA TTGTGAG CTGTAAC CTGTGAC ATGAGAC PWM GTGTTAA CTGTGAA TTGTGAG ATTTGAA TTGTGAC
58 Models based on Information Entropy Given a set of genomic sequences, it can be used to rank each member sequence according to its information content. This helps in evaluating each input sequence and takes care of weak signals which were completely ignored by earlier PWM model. Limitations The model based on information entropy measures the information quantity. It says nothing about the quality of information. Given two substrings in a sequence with equal quantity of information, only may contain biologically relevant information (e.g. motif).
59 Proposed Methodology : Shannon Entropy along with Kullback-Leibler Divergence Motivations Biological observation The biological motif, more stable structure and is expected to have lower entropy Physical observation Chaotic systems have a higher entropy value and the stable systems have lower entropy value. Motifs are short conserved regions in DNA, RNA, or Protein sequences and are expected to have lower entropies.
60 Proposed Methodology : Shannon Entropy Along With Kullback-Leibler Divergence From Kullback Leibler divergence In probability theory the Kullback Leibler divergence is a non-commutative measure of the difference between two probability distributions. It is non-negative and is 0 if both distributions are equivalent (p=q). The smaller the divergence, the more similar is the distribution of the two variables. This motivates that the measure based on KL divergence can be used to model the important aspects of interesting signals or significant patterns (e.g. motifs) in DNA sequences
61 Algorithm Step1. Given a set of sequences and length of an unknown signal, divide each sequence to get substrings of length equal to the given length of the signal. Step2. Compute the information entropy of all substrings obtained in Step 1. Step3. Choose those substrings that fall within a selected range of lower information entropies computed in step2. Step4. Find a consensus sequence of the set of substrings obtained in step3. Step5. Calculate the Kullback Leibler divergence for each substrings of step3 with respect to the consensus string obtained in step4. Step6. Keep those substrings that fall within a selected range of lower Kullback-Leibler divergence computed in step5. Step7. The consensus of the set of strings described in Step6 is the required signal.
62 Implementation We tested our method on various synthetic datasets. Randomly implanted motif As the results on the synthetic data set are promising, we applied our method to real data sets: transgenic data regions of E.Coli ECRDB70 database. Implemented in MatLab 7.1, Windows environment
63 Performance Evaluation The performance of our model on the real data set was computed using the following standard parameters Sensitivity: It is the percent of true motifs correctly predicted as true motifs. It measures how good our model in hitting the real motifs is. Specificity: It is the percent of non-motifs correctly predicted as non-motifs. It measures how good our model in hitting the non motifs. Accuracy: It is percentage of correctly predicted motifs. It measures how much accurate is the model in correctly predicting the motifs.
64 Result
65 Result Information Entropy Synthetic Data with randomly implanted motif Motif - Predicted - TAGCTTCATCGTTGA AAAAAAGAAAAAGAA
66 Result Kullback Leibler Divergence Synthetic Data with randomly implanted motif Motif AGAGAGAGAGAGAGA Predicted - AGAGAGAGAGAGAGA
67 Result Kullback Leibler Divergence Synthetic Data with randomly implanted motif Motif - TAGCTTCATCGTTGA Predicted - AATTTTCTTCATTTA
68 Result Kullback Leibler Divergence Synthetic Data with randomly implanted motif Motif - TAGCTTCATCGTTGA Predicted - TAGTTTCATCATTGA

69 Result Kullback Leibler Divergence E.Coli ECRDB Database Motif - AAATTTCGA Predicted - AACTCTCGA
70 Result The KL-Divergence model generated the consensus with an improved accuracy of 74% as compared to 57% in the model based only on Information entropy. The sensitivity and specificity of the KL- Divergence model were 75% and 72% respectively as compared to 33% and 59% in the model based on Information entropy.
71 Conclusion The PWM based model ignores the weak signals since the model completely discards the bases with lower probability of occurrence. The model based on information entropy overcomes the deficiencies associated with the PWM model but fails to distinguish between signal and noise when the information contents are same. The proposed model based on information entropy along with Kullback-Leibler divergence successfully addresses the problem of distinguishing between the relevant and irrelevant information.
72 conclusion We observe from the above test runs of the proposed algorithm that besides predicting the motifs which are correctly predicted by other methods it also predicts more effectively the motifs which are otherwise not correctly predicted by other models.
73 Motif Search in DNA Sequences Using Generalized Suffix Tree IEEE Computer Society,( ISBN: , DOI: / ICIT )
74 Generalized Suffix Tree Generalized Suffix tree (GST) When a set of strings are required to be represented by suffix tree, a generalized suffix tree (GST) is used. A GST for strings S1, S2, S3..SN can be built in O( S1 + S2 + S3 + SN ) time using Ukkonen algorithm.
75 Generalized suffix tree (Example) Let s 1 =abab and s 2 =aab here is a generalized suffix tree for s 1 and s 2 { } $ # b$ b# ab$ ab# bab$ aab# abab$ a b $ 1 b $ 3 # a 2 b a b # 1 a b$ $ 2 # $ 4 # 5 3 4
76 Related Work Leaf count information has been proven to be useful in reducing the search space for a given set of sequences. When the nodes are ranked on the basis of leaf count in a generalized suffix tree (GST) and if there is a partial match in the label of the first edge, the chances of full match in the subtree are highest. If there is no partial match, the biggest subtree from the search space can be eliminated from further processing. Limitations This may not always be true in biological applications. The occurrences of the motif in the given genomic sequences may not be exact. Hence it is important to find them even in the presence of small errors.
77 Contd Appropriate branching decision Reduce the search space. The branching decision is based on very limited information about the likelihood that a particular branch will lead to a solution. Limitation This decision is usually taken at a very early stage of the tree, yet it can have dominating influence on the overall efficiency of the search process. It may so happen that even the reasonable choices made at an early stage can lead to extremely poor results. Dynamic branching has been used as another strategy for further reduction of search space. It is capable of identifying un-influential branches on the basis of more reliable information available from later stages. Limitation It does not allow mismatches.
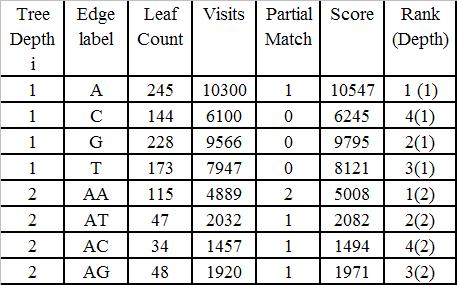
78 Additional Parameters Edge label length The number of characters on the edge from the root to a node, is an important parameter in measuring the biological information it carries. Frequency of visits of an edge More visits of an edge in the GST points to a more probable conserved region and signify higher probability of occurrences of that pattern in the motif. Partial match with interspersed mismatch During the process of exact matching if there is a partial match in an edge then the chances of a full match in the later stage is maximized.
79 Proposed Scoring Scheme Let x be the number of leaves in the subtree rooted at v. Edge length of node v is the number of characters from the root to v. Let t be edge length of node v, y be the frequency of visit of that edge. Let z be the number of matches of the edge label with the given motif with match weight w. Then the total score of node v = x + y*t + z*w.
80 Algorithm 1. Read all the input DNA sequences 2. Construct a generalized suffix tree (GST) for the input sequences. While constructing the GST, at each internal node at tree depth i, store the edge label length, number of leaves and frequency of visit of an edge. 3. For each edge label at tree depth i compute the partial match score comparing with the given motif. 4. Compute the scoring function for each of the nodes at tree depth i. Rank the nodes in nonincreasing order of the scoring function. If total score of two sub trees are equal we assign higher rank to the one that has more leaf count.
81 Algorithm. 5. Search the sub tree under that node that has got highest score Update l as l - number of characters in the path traversed so far. Update d as d - the number of mismatches in the path traversed so far. If along an edge number of mismatches >d then visit the next ranked node. 6. Otherwise 7. Repeat step 3 and 4 till a motif is reported. (l=0)
82 Result
83 Conclusion We have discussed the motif finding problem as a type of tree based search problem. Presented an algorithm that preprocess the generalized suffix tree using parameters like leaf counts, edge label length with frequency of its visit and partial match with allowable interspersed mismatches. A composite scoring scheme is used to rank the nodes and is used to discard the unproductive search space by selecting the highest ranking search space.
84 conclusion Allows mismatches Reduces search space Explained with synthetic and implanted data
85 Motif Prediction using Weighted Degree String Kernels with Shift and Mismatch. ACM (ICAC3-09), Pages 56-61, ACM ( ISBN: DOI:
86 Problem: Find a given motif of length l with up to m number of mismatches in a given set of DNA sequences. We use a class of string kernel, called weighted degree kernel with shift and extend it to allow mismatches. we use the suffix tree based mismatch tree data structure to train the SVM using a scoring scheme as a speedup measure during implementation
87 Support Vector Machines (SVM) Support Vector Machines (SVMs) are supervised learning algorithms first introduced by Vapnik in SVM use Kernel functions as the core for classification and regression analysis. The kernel function corresponds to an inner product in some expanded feature space. The computation in high dimensional feature space is avoided through the use of kernels. Higher dimensional feature space allows obtaining a linear separation for data which are otherwise not linearly separable in lower dimension.
88 Support Vector Machines (SVM)
89 Support Vector Machines (SVM) SVMs and related kernel methods are extremely good at solving prediction problems in computational biology. Can easily handle non-vector inputs such as variable length sequences or graphs. Ability to deal with high-dimensional large data sets. Flexibility in modeling even with small training samples. Due to Mercer s conditions on the kernels the corresponding optimization problems are convex and hence have no local minima. These facts mark a clear advantage of SVM over other pattern recognition algorithms, such as neural networks.
90 Related Work Weighted Degree Kernel [Ratsch, G., & Sonnenburg, S. (2004)] The WD kernel of order K compares two sequences x and x of equal length L by summing all contributions of k- mer matches of length k ε {1, 2,.K )weighted by coefficient β k ' K L k + 1 ' K ( x, x ) = βk I ( uk i ( x) = u k = 1 i= 1, k, i ' ( x )) where u k,i (x) = string of length k starting at position i of the sequence x. I(.) evaluates to 1 when the argument otherwise. ' 2( K k + 1) β k = ' ' K ( K + 1) is true, zero Limitations Recognition of matching blocks using WD kernel strongly depends on the position of the subsequence and does not tolerate any positional variation, e.g. if there is a single shift in one of the sequence by just only one position, the WD kernel fails to discover similar blocks and returns a lower similarity score
91 Related Work Weighted Degree Kernel with shift (WDS) [Ratsch, G., Sonnenburg, S., & Scholkopf, B. (2005)] WD kernel with shift tolerates small positional variations in the sequences to be compared. Where δ s =1 / 2(s+ 1) K(x,x ' ) = K ' k = 1 β k L k + 1 i= 1 s s= 0 s+ i L 2(K' k + 1) β k = K '(k+ 1) δ s μ k,i,s,x,x ' μ [ ' ' I(u (x) = u (x )) + I(u (x) u (x ))] = k,i,s,x,x ' k,i+ s k,i k,i = k,i+ s Limitations Do not allow mutations (mismatches)
92 Proposed Kernel To allow mutations in the candidate motifs, weighted degree kernel with shift and mismatches is proposed. Where ' K M k= 1m= 0 L k+ 1 i= 1 k( x, x ) = β δ μ μ ' k,i,s,x,x = I(u k,i + k, m (x) s m s s= 0 s+ i L ' u (x k,i k, i, s, x, x u u evaluated to true if and only if there are exactly m mismatches between u and u. Choose β k,m = total weight / no. of possible m-mismatching k-mer. s ) + I(u (x) k,i ' m u k,i + ' (x s )
93 Proposed Kernel To allow mutations in the candidate motifs, weighted degree kernel with shift and mismatches is proposed. ' K( x, x ) = β δ μ Where μ ' k,i,s,x,x = K ' M k= 1m= 0 I(u k,i + k, m (x) s L k+ 1 i= 1 m s s= 0 s+ i L ' u (x k,i k, i, s, x, x u u evaluated to true if and only if there are exactly m mismatches between u and u. Choose β k,m = total weight / no. of possible m-mismatching k-mer. ) s + I(u (x) k,i ' m u k,i + ' (x s )
94 Algorithm Step1: Step 2: Input the m training sequences and n test sequences from files in FASTA format Create two arrays of strings, one for the training DNA sequences and other for the test and validation DNA sequences. The sizes of these arrays are m x m and n x n respectively.
95 Step 3: Step 4: Initialize the parameters such as the length of the motif, allowable mismatches, maximum shift, and length of sequences to a desired value. Also initialize the constant C>0,that sets the relative importance of maximizing the margin and minimizing the amount of slack. Compute the kernel function for each pair of training sequences using the following steps: Step (4a): Compare all pairs of sequences by summing all contribution of the k-mer matches of length 1 to k.
96 Step (4b): Compute the indicator function which evaluates to 1 when the contributions are same, zero otherwise. Step (4c): Reward each matching substring by a weighting coefficient : β k β k, m β k,m = ( k m )( Σ 1 ) m Step 5: Step 6: Set the number of iterations, initial margin of separation and precision for the SVM computation, and initial values of the Lagrange multipliers α i Compute the optimum values for all the training sequences by solving the quadratic optimization problem using step 1 of the algorithm for general SVM.
97 Step 7: Step 8: Step 9: Compute the bias b using step 2 of the algorithm for general SVM. For each of the test and validation sequences compute the SVM decision function based on the computed parameters using step3 of the algorithm for general SVM. The value indicates whether the sequence contains the motif or not. Compute the performance measures.
98 Performance Evaluation Receiver operating Characteristic Curve (ROC Curve) The ROC is represented by plotting the true positive rate (TPR) vs. false positive rate (FPR). Area under the ROC curves (auroc): The area under Receiver Operating Curve is calculated by adding trapezoids. auroc quantifies the quality of the classifier and value >0.5 indicates better performance. Area under the PRC (auprc): PRC point = true positives / all true labels, true positives / (true positives + false positives) Cross Correlation coefficient (CC) CC = (true positives * true negatives - false positives * false negatives) / sqrt ( (true positives + false positives) * (true positives + false negatives) * (true negatives + false positives) *(true negatives + false negatives) ) F-Measure and Accuracy F-measure = 2 / (1 / precision + 1 / recall) Accuracy = (true positives + true negatives) / all labels
99 Implementation The proposed kernel was first tested on various synthetic data sets. As the results of these tests are promising, the kernel was applied to real data sets comprising of the C. Elegans and Drosophila Melanogaster acceptor sites. The SHOGUN tool box was used for implementation of the proposed kernel. The kernel was developed using c++. A suffix tree based data structure with a scoring scheme is used to speedup the SVM training and classification. Synthetic Data. randomly generated strings with signals of different length implanted at random positions Real Dataset. C.elegans acceptor sites, sequences containing true splice sites. (5.9%) Drosophila acceptor sites, sequences containing 1583 true splice sites. (1.6%)
100 Result Synthetic Data Synthetic Data (5 Mut)
101 Synthetic Data (5Mut)
102 Result Drosophila C.Elegans
103 Result
104 conclusion The WDSM kernel allows mismatches to be used along with shift while computing the kernel from the given DNA sequences. The mismatch and shift values together resulted in better representation of the motifs as well as the positions within the sequence. This has resulted in higher accuracy in classifying the DNA sequences for predicting the motifs. The method has ability to detect motifs even with a small size of training samples. A suffix tree based data structure with a scoring scheme is used to speedup the SVM training and classification.
105 Prediction of Motifs with Unknown Length and Allowable Mismatches Using Kernel Based Approach
106 Related Works Spectrum Kernel [Leslie, C., Kuang, R., & Eskin, E. (2003)] Especially tuned to small alphabets (like the 2-bit DNA alphabet). Defined as the inner product K( x, x') = Φ k ( x). Φ ( x') Where, Ø k maps a sequence x that consists of characters in to a feature space of size k Let #u(x) is the number of occurrence of u in x, k be the length of pattern u, be an alphabet Then Ø k (x) can be defined as φk( x) = (# u( x)) u ε k k
107 Related Works Weighted Degree kernel without shifts (WDKWS) introduced by Sonnenburg et al., 2006, uses a weighted sum of spectrum kernels for all degrees at every position of the sequence. The difference between the WDKWS and the spectrum kernel are the position dependence and the consideration of K-mers vs. 1 K-mers, i.e. i.e. also considering subsequences of the K-mers during computation.
108 Related Works Weighted Degree kernel with shift and mismatch (WDKSM) As already presented, aims at improving the representations of the weighted degree kernel which allows mismatches. Has resulted in higher accuracy in classifying the DNA sequences for predicting the motifs. Ability to detect motifs even with small sized training samples.
109 Limitations Spectrum Kernels Cannot extract any positional information from the sequence which goes beyond the k-mer length. Well suited for describing the content but not for analyzing signals where motifs appear in a certain order. Weighted Degree kernel without shifts When using long K-mers, the memory demand becomes intractable and the sparse weight vectors need to be stored and operated with more efficiently. Hence it is difficult to implement this kernel for practical purpose.
110 Limitations Weighted Degree kernel with shift and mismatch (WDKSM) This works fine for classification and finding motifs when the length of the motif and the number of allowable mismatches are known. Most motif discovery algorithms for DNA sequences require the motif's length as input. Styczynski et al. introduced the Extended (l,d)-motif Problem (EMP) where the motif's length is not an input parameter. Their algorithm takes an unacceptably long time to run.
111 Motivation for new Kernel Feature Space The size of the feature map Ø for string kernels for any l length pattern over alphabet A is of dimension l A. Feature space size increases exponentially with the increase in the pattern length. So difficult for higher length patterns. Most of the dimensions of the feature space are sparse. The feature space can be reduced to the total number of sub sequences occurring in the training set. Behavior of Spectrum Kernels the output no longer depends on the characteristics of the input such as from which organism the sequence has been obtained, the length of the sequence, the distribution of nucleotides etc. e.g. the kernel computed from AAAA, CCCC or AAAA, TTTT or GGGG, CCCC are same.
112 Motivation for new Kernel Local and global requirements of motif prediction The string kernels rely on the indicator functions and pair wise string comparisons for computation of the kernel value. Values should be more significant for related or similar sequences as compared to unrelated or dissimilar ones. Not observed in the existing string kernels as the sequence comparisons are made starting from single nucleotides to a certain length, allowing shifts and mismatches We have proposed a kernel which provides meaningful scores depending only on the motifs it contain The kernel abstraction can be utilized for designing a generalized kernel for detection of unknown motifs without depending on the training information of the organisms or other domain specific information.
113 Motivation for new Kernel Design a new spectrum kernel to possibly cater to the above observations by reducing search space, generating meaningful values which can be generalized.
114 Proposed Weighted Spectrum Kernel Let S be a set of n sequences each of length l from alphabet for which we intend to determine the motifs with unknown length l and known mutations d. Let x and x be pair of sequences from S, u i be any k- mer of length k occurring in x at position i and u i (x,x ) be the number of occurrences of u i in x and x. Let u i (S-{x,x }) be the number of occurrences of u i in the sequences of the training set S-{x,x }, B i be the weight attached to each position i. The weight coefficient B i = 2(T- u i +1)/T(T+1), where T is the total number of all possible length k k-mers occurring in the training set S-{x,x }.
115 Proposed Weighted Spectrum Kernel Then the proposed weighted degree spectrum kernel is defined as: K( x, x') = l k+ 1 B i u ( x, x i= 1 { x, x'}) This kernel during computation of the spectrum values besides considering the number of times the pattern occurring in the pair of sequences being compared, also considers the number of times the k- mer occurs in the other sequences of the training set. i ' ) u i ( S
116 Implementation The proposed kernel was first tested on various synthetic data sets. As the results of these tests are promising, the kernel was applied to real data sets comprising of the C. Elegans and Drosophila Melanogaster acceptor sites and also the transcription start sites (TSS) of human genome hg16. The kernel was developed using c++. The SHOGUN tool box was used for implementation of the proposed kernel for the quadratic optimization. Synthetic Data. randomly generated strings with signals of different length implanted at random positions. Real Dataset. C.elegans acceptor sites, sequences containing true splice sites. (5.9%) Drosophila acceptor sites, sequences containing 1583 true splice sites. (1.6%) Hg16 TSS of human genome with 14,18,392 sequences containing true sites (1.05%)
117 Result
118 Result
119 Result
120 Result
121 Result
122 Result Synthetic data set 1 Kernel Accuracy AuROC Fixed Degree 65.00% Spectrum Kernel 38.00% Weighted String Kernel 53.00% Weighted Spectrum Kernel Synthetic data set % Kernel Accuracy AuROC Fixed Degree 90.15% Spectrum Kernel 89.23% Weighted String Kernel 90.90% Weighted Spectrum Kernel 90.95% 0.803
123 Result C.Elegans Acceptor data Kernel Accuracy AuROC Fixed Degree 93.10% 0.85 Spectrum Kernel 94.05% 0.57 Weighted String Kernel 92.10% 0.63 Weighted Spectrum Kernel 94.10% 0.91
124 Result Drosophila Acceptor data Kernel Accuracy AuROC Fixed Degree 98.39% Spectrum Kernel 97.55% Weighted String Kernel 94.79% Weighted Spectrum Kernel 98.39% 0.881
125 Result Hg16 TSS data Kernel Accuracy AuROC Fixed Degree 96.94% 0.66 Spectrum Kernel 97.33% 0.59 Weighted String Kernel 97.10% 0.71 Weighted Spectrum Kernel 98.94% 0.75
126 conclusions The performance of the new kernel was found to be better both in terms of the classification accuracy and auroc. The kernel also outperformed the existing string kernels by allowing for classification with motif length of more than 8. The new kernel yielded better performance from the other kernels with a considerable improvement of the auroc from to improved performance of the SVM classifier using the new kernel was also observed when the data sets containing acceptor sites of Drosophila, C.Elegans were used. the ability of the SVM to classify sequences with mutated motifs using the same training data. The accuracy of classification did not depend on the training motif, the mutations and to some extent on the length.
127 conclusion Cross classification of sequences for prediction of unknown motif. The SVM acceptor sites of drosophila was used to predict the C.Elegans acceptor sites and showed an accuracy of 94% similar to the result obtained when trained with its own samples.
128 References Leslie, C., Kuang, R.,and Eskin, E., Inexact matching string kernels for protein classification, Kernel Methods in Computational Biology,pp , MIT Press, Ratsch, G., Sonnenburg, S., & Scholkopf, B. (2005). Rase: Recognition of alternatively spliced exons in c. elegans. ISMB Ratsch, G., & Sonnenburg, S. (2004). Accurate splice site prediction for caenorhabditis elegans, MIT Press series on Computational Molecular Biology. MIT Press. Sonnenburg, S., Raetsch, G., Schaefer, C.,and Schoelkopf, B., Large Scale Multiple Kernel Learning, Journal of Machine Learning Research,7, pp , Styczynski, M.P., Jensen, K.L., Rigoutsos, I.,and Stephanopoulos, G.N., An extension and novel solution to the (l,d)-motif challenge problem, Genome Informatics, Vol. 15, pp.63 71, 2004.
129 THANK YOU
Practical Bioinformatics
 5/2/2017 Dictionaries d i c t i o n a r y = { A : T, T : A, G : C, C : G } d i c t i o n a r y [ G ] d i c t i o n a r y [ N ] = N d i c t i o n a r y. h a s k e y ( C ) Dictionaries g e n e t i c C o
5/2/2017 Dictionaries d i c t i o n a r y = { A : T, T : A, G : C, C : G } d i c t i o n a r y [ G ] d i c t i o n a r y [ N ] = N d i c t i o n a r y. h a s k e y ( C ) Dictionaries g e n e t i c C o
How can one gene have such drastic effects?
 Slides revised and adapted Computational Biology course IST Ana Teresa Freitas 2011/2012 A recent microarray experiment showed that when gene X is knocked out, 20 other genes are not expressed How can
Slides revised and adapted Computational Biology course IST Ana Teresa Freitas 2011/2012 A recent microarray experiment showed that when gene X is knocked out, 20 other genes are not expressed How can
9/8/09 Comp /Comp Fall
 9/8/09 Comp 590-90/Comp 790-90 Fall 2009 1 Genomes contain billions of bases (10 9 ) Within these there are 10s of 1000s genes (10 4 ) Genes are 1000s of bases long on average (10 3 ) So only 1% of DNA
9/8/09 Comp 590-90/Comp 790-90 Fall 2009 1 Genomes contain billions of bases (10 9 ) Within these there are 10s of 1000s genes (10 4 ) Genes are 1000s of bases long on average (10 3 ) So only 1% of DNA
SEQUENCE ALIGNMENT BACKGROUND: BIOINFORMATICS. Prokaryotes and Eukaryotes. DNA and RNA
 SEQUENCE ALIGNMENT BACKGROUND: BIOINFORMATICS 1 Prokaryotes and Eukaryotes 2 DNA and RNA 3 4 Double helix structure Codons Codons are triplets of bases from the RNA sequence. Each triplet defines an amino-acid.
SEQUENCE ALIGNMENT BACKGROUND: BIOINFORMATICS 1 Prokaryotes and Eukaryotes 2 DNA and RNA 3 4 Double helix structure Codons Codons are triplets of bases from the RNA sequence. Each triplet defines an amino-acid.
Advanced topics in bioinformatics
 Feinberg Graduate School of the Weizmann Institute of Science Advanced topics in bioinformatics Shmuel Pietrokovski & Eitan Rubin Spring 2003 Course WWW site: http://bioinformatics.weizmann.ac.il/courses/atib
Feinberg Graduate School of the Weizmann Institute of Science Advanced topics in bioinformatics Shmuel Pietrokovski & Eitan Rubin Spring 2003 Course WWW site: http://bioinformatics.weizmann.ac.il/courses/atib
Finding Regulatory Motifs in DNA Sequences
 Finding Regulatory Motifs in DNA Sequences Outline Implanting Patterns in Random Text Gene Regulation Regulatory Motifs The Gold Bug Problem The Motif Finding Problem Brute Force Motif Finding The Median
Finding Regulatory Motifs in DNA Sequences Outline Implanting Patterns in Random Text Gene Regulation Regulatory Motifs The Gold Bug Problem The Motif Finding Problem Brute Force Motif Finding The Median
Finding Regulatory Motifs in DNA Sequences
 Finding Regulatory Motifs in DNA Sequences Outline Implanting Patterns in Random Text Gene Regulation Regulatory Motifs The Gold Bug Problem The Motif Finding Problem Brute Force Motif Finding The Median
Finding Regulatory Motifs in DNA Sequences Outline Implanting Patterns in Random Text Gene Regulation Regulatory Motifs The Gold Bug Problem The Motif Finding Problem Brute Force Motif Finding The Median
Finding Regulatory Motifs in DNA Sequences
 Finding Regulatory Motifs in DNA Sequences Outline Implanting Patterns in Random Text Gene Regulation Regulatory Motifs The Gold Bug Problem The Motif Finding Problem Brute Force Motif Finding The Median
Finding Regulatory Motifs in DNA Sequences Outline Implanting Patterns in Random Text Gene Regulation Regulatory Motifs The Gold Bug Problem The Motif Finding Problem Brute Force Motif Finding The Median
Algorithms for Bioinformatics
 These slides use material from http://bix.ucsd.edu/bioalgorithms/slides.php 582670 Algorithms for Bioinformatics Lecture 2: Exhaustive search and motif finding 6.9.202 Outline Implanted motifs - an introduction
These slides use material from http://bix.ucsd.edu/bioalgorithms/slides.php 582670 Algorithms for Bioinformatics Lecture 2: Exhaustive search and motif finding 6.9.202 Outline Implanted motifs - an introduction
Algorithms for Bioinformatics
 These slides are based on previous years slides by Alexandru Tomescu, Leena Salmela and Veli Mäkinen These slides use material from http://bix.ucsd.edu/bioalgorithms/slides.php 582670 Algorithms for Bioinformatics
These slides are based on previous years slides by Alexandru Tomescu, Leena Salmela and Veli Mäkinen These slides use material from http://bix.ucsd.edu/bioalgorithms/slides.php 582670 Algorithms for Bioinformatics
Supplementary Information for
 Supplementary Information for Evolutionary conservation of codon optimality reveals hidden signatures of co-translational folding Sebastian Pechmann & Judith Frydman Department of Biology and BioX, Stanford
Supplementary Information for Evolutionary conservation of codon optimality reveals hidden signatures of co-translational folding Sebastian Pechmann & Judith Frydman Department of Biology and BioX, Stanford
Crick s early Hypothesis Revisited
 Crick s early Hypothesis Revisited Or The Existence of a Universal Coding Frame Ryan Rossi, Jean-Louis Lassez and Axel Bernal UPenn Center for Bioinformatics BIOINFORMATICS The application of computer
Crick s early Hypothesis Revisited Or The Existence of a Universal Coding Frame Ryan Rossi, Jean-Louis Lassez and Axel Bernal UPenn Center for Bioinformatics BIOINFORMATICS The application of computer
SUPPORTING INFORMATION FOR. SEquence-Enabled Reassembly of β-lactamase (SEER-LAC): a Sensitive Method for the Detection of Double-Stranded DNA
: a Sensitive Method for the Detection of Double-Stranded DNA") SUPPORTING INFORMATION FOR SEquence-Enabled Reassembly of β-lactamase (SEER-LAC): a Sensitive Method for the Detection of Double-Stranded DNA Aik T. Ooi, Cliff I. Stains, Indraneel Ghosh *, David J. Segal
SUPPORTING INFORMATION FOR SEquence-Enabled Reassembly of β-lactamase (SEER-LAC): a Sensitive Method for the Detection of Double-Stranded DNA Aik T. Ooi, Cliff I. Stains, Indraneel Ghosh *, David J. Segal
High throughput near infrared screening discovers DNA-templated silver clusters with peak fluorescence beyond 950 nm
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2018 High throughput near infrared screening discovers DNA-templated silver clusters with peak fluorescence
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2018 High throughput near infrared screening discovers DNA-templated silver clusters with peak fluorescence
Supplemental data. Pommerrenig et al. (2011). Plant Cell /tpc
. Plant Cell /tpc") Supplemental Figure 1. Prediction of phloem-specific MTK1 expression in Arabidopsis shoots and roots. The images and the corresponding numbers showing absolute (A) or relative expression levels (B) of
Supplemental Figure 1. Prediction of phloem-specific MTK1 expression in Arabidopsis shoots and roots. The images and the corresponding numbers showing absolute (A) or relative expression levels (B) of
6.047 / Computational Biology: Genomes, Networks, Evolution Fall 2008
 MIT OpenCourseWare http://ocw.mit.edu 6.047 / 6.878 Computational Biology: Genomes, Networks, Evolution Fall 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms.
MIT OpenCourseWare http://ocw.mit.edu 6.047 / 6.878 Computational Biology: Genomes, Networks, Evolution Fall 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms.
Characterization of Pathogenic Genes through Condensed Matrix Method, Case Study through Bacterial Zeta Toxin
 International Journal of Genetic Engineering and Biotechnology. ISSN 0974-3073 Volume 2, Number 1 (2011), pp. 109-114 International Research Publication House http://www.irphouse.com Characterization of
International Journal of Genetic Engineering and Biotechnology. ISSN 0974-3073 Volume 2, Number 1 (2011), pp. 109-114 International Research Publication House http://www.irphouse.com Characterization of
Regulatory Sequence Analysis. Sequence models (Bernoulli and Markov models)
") Regulatory Sequence Analysis Sequence models (Bernoulli and Markov models) 1 Why do we need random models? Any pattern discovery relies on an underlying model to estimate the random expectation. This model
Regulatory Sequence Analysis Sequence models (Bernoulli and Markov models) 1 Why do we need random models? Any pattern discovery relies on an underlying model to estimate the random expectation. This model
Clay Carter. Department of Biology. QuickTime and a TIFF (Uncompressed) decompressor are needed to see this picture.
 decompressor are needed to see this picture.") QuickTime and a TIFF (Uncompressed) decompressor are needed to see this picture. Clay Carter Department of Biology QuickTime and a TIFF (LZW) decompressor are needed to see this picture. Ornamental tobacco
QuickTime and a TIFF (Uncompressed) decompressor are needed to see this picture. Clay Carter Department of Biology QuickTime and a TIFF (LZW) decompressor are needed to see this picture. Ornamental tobacco
Algorithmische Bioinformatik WS 11/12:, by R. Krause/ K. Reinert, 14. November 2011, 12: Motif finding
 Algorithmische Bioinformatik WS 11/12:, by R. Krause/ K. Reinert, 14. November 2011, 12:00 4001 Motif finding This exposition was developed by Knut Reinert and Clemens Gröpl. It is based on the following
Algorithmische Bioinformatik WS 11/12:, by R. Krause/ K. Reinert, 14. November 2011, 12:00 4001 Motif finding This exposition was developed by Knut Reinert and Clemens Gröpl. It is based on the following
Partial restriction digest
 This lecture Exhaustive search Torgeir R. Hvidsten Restriction enzymes and the partial digest problem Finding regulatory motifs in DNA Sequences Exhaustive search methods T.R. Hvidsten: 1MB304: Discrete
This lecture Exhaustive search Torgeir R. Hvidsten Restriction enzymes and the partial digest problem Finding regulatory motifs in DNA Sequences Exhaustive search methods T.R. Hvidsten: 1MB304: Discrete
Nature Structural & Molecular Biology: doi: /nsmb Supplementary Figure 1
 Supplementary Figure 1 Zn 2+ -binding sites in USP18. (a) The two molecules of USP18 present in the asymmetric unit are shown. Chain A is shown in blue, chain B in green. Bound Zn 2+ ions are shown as
Supplementary Figure 1 Zn 2+ -binding sites in USP18. (a) The two molecules of USP18 present in the asymmetric unit are shown. Chain A is shown in blue, chain B in green. Bound Zn 2+ ions are shown as
Number-controlled spatial arrangement of gold nanoparticles with
 Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2016 Number-controlled spatial arrangement of gold nanoparticles with DNA dendrimers Ping Chen,*
Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2016 Number-controlled spatial arrangement of gold nanoparticles with DNA dendrimers Ping Chen,*
Modelling and Analysis in Bioinformatics. Lecture 1: Genomic k-mer Statistics
 582746 Modelling and Analysis in Bioinformatics Lecture 1: Genomic k-mer Statistics Juha Kärkkäinen 06.09.2016 Outline Course introduction Genomic k-mers 1-Mers 2-Mers 3-Mers k-mers for Larger k Outline
582746 Modelling and Analysis in Bioinformatics Lecture 1: Genomic k-mer Statistics Juha Kärkkäinen 06.09.2016 Outline Course introduction Genomic k-mers 1-Mers 2-Mers 3-Mers k-mers for Larger k Outline
SSR ( ) Vol. 48 No ( Microsatellite marker) ( Simple sequence repeat,ssr),
 Vol. 48 No ( Microsatellite marker) ( Simple sequence repeat,ssr),") 48 3 () Vol. 48 No. 3 2009 5 Journal of Xiamen University (Nat ural Science) May 2009 SSR,,,, 3 (, 361005) : SSR. 21 516,410. 60 %96. 7 %. (),(Between2groups linkage method),.,, 11 (),. 12,. (, ), : 0.
48 3 () Vol. 48 No. 3 2009 5 Journal of Xiamen University (Nat ural Science) May 2009 SSR,,,, 3 (, 361005) : SSR. 21 516,410. 60 %96. 7 %. (),(Between2groups linkage method),.,, 11 (),. 12,. (, ), : 0.
Electronic supplementary material
 Applied Microbiology and Biotechnology Electronic supplementary material A family of AA9 lytic polysaccharide monooxygenases in Aspergillus nidulans is differentially regulated by multiple substrates and
Applied Microbiology and Biotechnology Electronic supplementary material A family of AA9 lytic polysaccharide monooxygenases in Aspergillus nidulans is differentially regulated by multiple substrates and
SUPPLEMENTARY DATA - 1 -
 - 1 - SUPPLEMENTARY DATA Construction of B. subtilis rnpb complementation plasmids For complementation, the B. subtilis rnpb wild-type gene (rnpbwt) under control of its native rnpb promoter and terminator
- 1 - SUPPLEMENTARY DATA Construction of B. subtilis rnpb complementation plasmids For complementation, the B. subtilis rnpb wild-type gene (rnpbwt) under control of its native rnpb promoter and terminator
NSCI Basic Properties of Life and The Biochemistry of Life on Earth
 NSCI 314 LIFE IN THE COSMOS 4 Basic Properties of Life and The Biochemistry of Life on Earth Dr. Karen Kolehmainen Department of Physics CSUSB http://physics.csusb.edu/~karen/ WHAT IS LIFE? HARD TO DEFINE,
NSCI 314 LIFE IN THE COSMOS 4 Basic Properties of Life and The Biochemistry of Life on Earth Dr. Karen Kolehmainen Department of Physics CSUSB http://physics.csusb.edu/~karen/ WHAT IS LIFE? HARD TO DEFINE,
Evolvable Neural Networks for Time Series Prediction with Adaptive Learning Interval
 Evolvable Neural Networs for Time Series Prediction with Adaptive Learning Interval Dong-Woo Lee *, Seong G. Kong *, and Kwee-Bo Sim ** *Department of Electrical and Computer Engineering, The University
Evolvable Neural Networs for Time Series Prediction with Adaptive Learning Interval Dong-Woo Lee *, Seong G. Kong *, and Kwee-Bo Sim ** *Department of Electrical and Computer Engineering, The University
Supplementary Information
 Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2014 Directed self-assembly of genomic sequences into monomeric and polymeric branched DNA structures
Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2014 Directed self-assembly of genomic sequences into monomeric and polymeric branched DNA structures
Algorithms for Bioinformatics
 These slides are based on previous years slides by Alexandru Tomescu, Leena Salmela and Veli Mäkinen These slides use material from http://bix.ucsd.edu/bioalgorithms/slides.php Algorithms for Bioinformatics
These slides are based on previous years slides by Alexandru Tomescu, Leena Salmela and Veli Mäkinen These slides use material from http://bix.ucsd.edu/bioalgorithms/slides.php Algorithms for Bioinformatics
Supporting Information
 Supporting Information T. Pellegrino 1,2,3,#, R. A. Sperling 1,#, A. P. Alivisatos 2, W. J. Parak 1,2,* 1 Center for Nanoscience, Ludwig Maximilians Universität München, München, Germany 2 Department of
Supporting Information T. Pellegrino 1,2,3,#, R. A. Sperling 1,#, A. P. Alivisatos 2, W. J. Parak 1,2,* 1 Center for Nanoscience, Ludwig Maximilians Universität München, München, Germany 2 Department of
Supplemental Table 1. Primers used for cloning and PCR amplification in this study
 Supplemental Table 1. Primers used for cloning and PCR amplification in this study Target Gene Primer sequence NATA1 (At2g393) forward GGG GAC AAG TTT GTA CAA AAA AGC AGG CTT CAT GGC GCC TCC AAC CGC AGC
Supplemental Table 1. Primers used for cloning and PCR amplification in this study Target Gene Primer sequence NATA1 (At2g393) forward GGG GAC AAG TTT GTA CAA AAA AGC AGG CTT CAT GGC GCC TCC AAC CGC AGC
Building a Multifunctional Aptamer-Based DNA Nanoassembly for Targeted Cancer Therapy
 Supporting Information Building a Multifunctional Aptamer-Based DNA Nanoassembly for Targeted Cancer Therapy Cuichen Wu,, Da Han,, Tao Chen,, Lu Peng, Guizhi Zhu,, Mingxu You,, Liping Qiu,, Kwame Sefah,
Supporting Information Building a Multifunctional Aptamer-Based DNA Nanoassembly for Targeted Cancer Therapy Cuichen Wu,, Da Han,, Tao Chen,, Lu Peng, Guizhi Zhu,, Mingxu You,, Liping Qiu,, Kwame Sefah,
Supplemental Figure 1.
 A wt spoiiiaδ spoiiiahδ bofaδ B C D E spoiiiaδ, bofaδ Supplemental Figure 1. GFP-SpoIVFA is more mislocalized in the absence of both BofA and SpoIIIAH. Sporulation was induced by resuspension in wild-type
A wt spoiiiaδ spoiiiahδ bofaδ B C D E spoiiiaδ, bofaδ Supplemental Figure 1. GFP-SpoIVFA is more mislocalized in the absence of both BofA and SpoIIIAH. Sporulation was induced by resuspension in wild-type
Supporting Information for. Initial Biochemical and Functional Evaluation of Murine Calprotectin Reveals Ca(II)-
-") Supporting Information for Initial Biochemical and Functional Evaluation of Murine Calprotectin Reveals Ca(II)- Dependence and Its Ability to Chelate Multiple Nutrient Transition Metal Ions Rose C. Hadley,
Supporting Information for Initial Biochemical and Functional Evaluation of Murine Calprotectin Reveals Ca(II)- Dependence and Its Ability to Chelate Multiple Nutrient Transition Metal Ions Rose C. Hadley,
TM1 TM2 TM3 TM4 TM5 TM6 TM bp
 a 467 bp 1 482 2 93 3 321 4 7 281 6 21 7 66 8 176 19 12 13 212 113 16 8 b ATG TCA GGA CAT GTA ATG GAG GAA TGT GTA GTT CAC GGT ACG TTA GCG GCA GTA TTG CGT TTA ATG GGC GTA GTG M S G H V M E E C V V H G T
a 467 bp 1 482 2 93 3 321 4 7 281 6 21 7 66 8 176 19 12 13 212 113 16 8 b ATG TCA GGA CAT GTA ATG GAG GAA TGT GTA GTT CAC GGT ACG TTA GCG GCA GTA TTG CGT TTA ATG GGC GTA GTG M S G H V M E E C V V H G T
Table S1. Primers and PCR conditions used in this paper Primers Sequence (5 3 ) Thermal conditions Reference Rhizobacteria 27F 1492R
 Thermal conditions Reference Rhizobacteria 27F 1492R") Table S1. Primers and PCR conditions used in this paper Primers Sequence (5 3 ) Thermal conditions Reference Rhizobacteria 27F 1492R AAC MGG ATT AGA TAC CCK G GGY TAC CTT GTT ACG ACT T Detection of Candidatus
Table S1. Primers and PCR conditions used in this paper Primers Sequence (5 3 ) Thermal conditions Reference Rhizobacteria 27F 1492R AAC MGG ATT AGA TAC CCK G GGY TAC CTT GTT ACG ACT T Detection of Candidatus
Algorithms in Bioinformatics II SS 07 ZBIT, C. Dieterich, (modified script of D. Huson), April 25,
, April 25,") Algorithms in Bioinformatics II SS 07 ZBIT, C. Dieterich, (modified script of D. Huson), April 25, 200707 Motif Finding This exposition is based on the following sources, which are all recommended reading:.
Algorithms in Bioinformatics II SS 07 ZBIT, C. Dieterich, (modified script of D. Huson), April 25, 200707 Motif Finding This exposition is based on the following sources, which are all recommended reading:.
Protein Threading. Combinatorial optimization approach. Stefan Balev.
 Protein Threading Combinatorial optimization approach Stefan Balev Stefan.Balev@univ-lehavre.fr Laboratoire d informatique du Havre Université du Havre Stefan Balev Cours DEA 30/01/2004 p.1/42 Outline
Protein Threading Combinatorial optimization approach Stefan Balev Stefan.Balev@univ-lehavre.fr Laboratoire d informatique du Havre Université du Havre Stefan Balev Cours DEA 30/01/2004 p.1/42 Outline
Encoding of Amino Acids and Proteins from a Communications and Information Theoretic Perspective
 Jacobs University Bremen Encoding of Amino Acids and Proteins from a Communications and Information Theoretic Perspective Semester Project II By: Dawit Nigatu Supervisor: Prof. Dr. Werner Henkel Transmission
Jacobs University Bremen Encoding of Amino Acids and Proteins from a Communications and Information Theoretic Perspective Semester Project II By: Dawit Nigatu Supervisor: Prof. Dr. Werner Henkel Transmission
SUPPLEMENTARY INFORMATION
 DOI:.8/NCHEM. Conditionally Fluorescent Molecular Probes for Detecting Single Base Changes in Double-stranded DNA Sherry Xi Chen, David Yu Zhang, Georg Seelig. Analytic framework and probe design.. Design
DOI:.8/NCHEM. Conditionally Fluorescent Molecular Probes for Detecting Single Base Changes in Double-stranded DNA Sherry Xi Chen, David Yu Zhang, Georg Seelig. Analytic framework and probe design.. Design
Pattern Matching (Exact Matching) Overview
 Overview") CSI/BINF 5330 Pattern Matching (Exact Matching) Young-Rae Cho Associate Professor Department of Computer Science Baylor University Overview Pattern Matching Exhaustive Search DFA Algorithm KMP Algorithm
CSI/BINF 5330 Pattern Matching (Exact Matching) Young-Rae Cho Associate Professor Department of Computer Science Baylor University Overview Pattern Matching Exhaustive Search DFA Algorithm KMP Algorithm
Evolutionary dynamics of abundant stop codon readthrough in Anopheles and Drosophila
 biorxiv preprint first posted online May. 3, 2016; doi: http://dx.doi.org/10.1101/051557. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder. All rights reserved.
biorxiv preprint first posted online May. 3, 2016; doi: http://dx.doi.org/10.1101/051557. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder. All rights reserved.
SUPPLEMENTARY INFORMATION
 SUPPLEMENTARY INFORMATION DOI:.38/NCHEM.246 Optimizing the specificity of nucleic acid hyridization David Yu Zhang, Sherry Xi Chen, and Peng Yin. Analytic framework and proe design 3.. Concentration-adjusted
SUPPLEMENTARY INFORMATION DOI:.38/NCHEM.246 Optimizing the specificity of nucleic acid hyridization David Yu Zhang, Sherry Xi Chen, and Peng Yin. Analytic framework and proe design 3.. Concentration-adjusted
3. Evolution makes sense of homologies. 3. Evolution makes sense of homologies. 3. Evolution makes sense of homologies
 Richard Owen (1848) introduced the term Homology to refer to structural similarities among organisms. To Owen, these similarities indicated that organisms were created following a common plan or archetype.
Richard Owen (1848) introduced the term Homology to refer to structural similarities among organisms. To Owen, these similarities indicated that organisms were created following a common plan or archetype.
Introduction to Molecular Phylogeny
 Introduction to Molecular Phylogeny Starting point: a set of homologous, aligned DNA or protein sequences Result of the process: a tree describing evolutionary relationships between studied sequences =
Introduction to Molecular Phylogeny Starting point: a set of homologous, aligned DNA or protein sequences Result of the process: a tree describing evolutionary relationships between studied sequences =
Why do more divergent sequences produce smaller nonsynonymous/synonymous
 Genetics: Early Online, published on June 21, 2013 as 10.1534/genetics.113.152025 Why do more divergent sequences produce smaller nonsynonymous/synonymous rate ratios in pairwise sequence comparisons?
Genetics: Early Online, published on June 21, 2013 as 10.1534/genetics.113.152025 Why do more divergent sequences produce smaller nonsynonymous/synonymous rate ratios in pairwise sequence comparisons?
evoglow - express N kit distributed by Cat.#: FP product information broad host range vectors - gram negative bacteria
 evoglow - express N kit broad host range vectors - gram negative bacteria product information distributed by Cat.#: FP-21020 Content: Product Overview... 3 evoglow express N -kit... 3 The evoglow -Fluorescent
evoglow - express N kit broad host range vectors - gram negative bacteria product information distributed by Cat.#: FP-21020 Content: Product Overview... 3 evoglow express N -kit... 3 The evoglow -Fluorescent
Support Vector Machines.
 Support Vector Machines www.cs.wisc.edu/~dpage 1 Goals for the lecture you should understand the following concepts the margin slack variables the linear support vector machine nonlinear SVMs the kernel
Support Vector Machines www.cs.wisc.edu/~dpage 1 Goals for the lecture you should understand the following concepts the margin slack variables the linear support vector machine nonlinear SVMs the kernel
Alignment. Peak Detection
 ChIP seq ChIP Seq Hongkai Ji et al. Nature Biotechnology 26: 1293-1300. 2008 ChIP Seq Analysis Alignment Peak Detection Annotation Visualization Sequence Analysis Motif Analysis Alignment ELAND Bowtie
ChIP seq ChIP Seq Hongkai Ji et al. Nature Biotechnology 26: 1293-1300. 2008 ChIP Seq Analysis Alignment Peak Detection Annotation Visualization Sequence Analysis Motif Analysis Alignment ELAND Bowtie
The 3 Genomic Numbers Discovery: How Our Genome Single-Stranded DNA Sequence Is Self-Designed as a Numerical Whole
 Applied Mathematics, 2013, 4, 37-53 http://dx.doi.org/10.4236/am.2013.410a2004 Published Online October 2013 (http://www.scirp.org/journal/am) The 3 Genomic Numbers Discovery: How Our Genome Single-Stranded
Applied Mathematics, 2013, 4, 37-53 http://dx.doi.org/10.4236/am.2013.410a2004 Published Online October 2013 (http://www.scirp.org/journal/am) The 3 Genomic Numbers Discovery: How Our Genome Single-Stranded
MCMC: Markov Chain Monte Carlo
 I529: Machine Learning in Bioinformatics (Spring 2013) MCMC: Markov Chain Monte Carlo Yuzhen Ye School of Informatics and Computing Indiana University, Bloomington Spring 2013 Contents Review of Markov
I529: Machine Learning in Bioinformatics (Spring 2013) MCMC: Markov Chain Monte Carlo Yuzhen Ye School of Informatics and Computing Indiana University, Bloomington Spring 2013 Contents Review of Markov
The Physical Language of Molecules
 The Physical Language of Molecules How do molecular codes emerge and evolve? International Workshop on Bio Soft Matter Tokyo, 2008 Biological information is carried by molecules Self replicating information
The Physical Language of Molecules How do molecular codes emerge and evolve? International Workshop on Bio Soft Matter Tokyo, 2008 Biological information is carried by molecules Self replicating information
On the Monotonicity of the String Correction Factor for Words with Mismatches
 On the Monotonicity of the String Correction Factor for Words with Mismatches (extended abstract) Alberto Apostolico Georgia Tech & Univ. of Padova Cinzia Pizzi Univ. of Padova & Univ. of Helsinki Abstract.
On the Monotonicity of the String Correction Factor for Words with Mismatches (extended abstract) Alberto Apostolico Georgia Tech & Univ. of Padova Cinzia Pizzi Univ. of Padova & Univ. of Helsinki Abstract.
part 3: analysis of natural selection pressure
 part 3: analysis of natural selection pressure markov models are good phenomenological codon models do have many benefits: o principled framework for statistical inference o avoiding ad hoc corrections
part 3: analysis of natural selection pressure markov models are good phenomenological codon models do have many benefits: o principled framework for statistical inference o avoiding ad hoc corrections
Topographic Mapping and Dimensionality Reduction of Binary Tensor Data of Arbitrary Rank
 Topographic Mapping and Dimensionality Reduction of Binary Tensor Data of Arbitrary Rank Peter Tiňo, Jakub Mažgút, Hong Yan, Mikael Bodén Topographic Mapping and Dimensionality Reduction of Binary Tensor
Topographic Mapping and Dimensionality Reduction of Binary Tensor Data of Arbitrary Rank Peter Tiňo, Jakub Mažgút, Hong Yan, Mikael Bodén Topographic Mapping and Dimensionality Reduction of Binary Tensor
Learning Sequence Motif Models Using Expectation Maximization (EM) and Gibbs Sampling
 and Gibbs Sampling") Learning Sequence Motif Models Using Expectation Maximization (EM) and Gibbs Sampling BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 009 Mark Craven craven@biostat.wisc.edu Sequence Motifs what is a sequence
Learning Sequence Motif Models Using Expectation Maximization (EM) and Gibbs Sampling BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 009 Mark Craven craven@biostat.wisc.edu Sequence Motifs what is a sequence
Using algebraic geometry for phylogenetic reconstruction
 Using algebraic geometry for phylogenetic reconstruction Marta Casanellas i Rius (joint work with Jesús Fernández-Sánchez) Departament de Matemàtica Aplicada I Universitat Politècnica de Catalunya IMA
Using algebraic geometry for phylogenetic reconstruction Marta Casanellas i Rius (joint work with Jesús Fernández-Sánchez) Departament de Matemàtica Aplicada I Universitat Politècnica de Catalunya IMA
evoglow - express N kit Cat. No.: product information broad host range vectors - gram negative bacteria
 evoglow - express N kit broad host range vectors - gram negative bacteria product information Cat. No.: 2.1.020 evocatal GmbH 2 Content: Product Overview... 4 evoglow express N kit... 4 The evoglow Fluorescent
evoglow - express N kit broad host range vectors - gram negative bacteria product information Cat. No.: 2.1.020 evocatal GmbH 2 Content: Product Overview... 4 evoglow express N kit... 4 The evoglow Fluorescent
Lecture 8 Learning Sequence Motif Models Using Expectation Maximization (EM) Colin Dewey February 14, 2008
 Colin Dewey February 14, 2008") Lecture 8 Learning Sequence Motif Models Using Expectation Maximization (EM) Colin Dewey February 14, 2008 1 Sequence Motifs what is a sequence motif? a sequence pattern of biological significance typically
Lecture 8 Learning Sequence Motif Models Using Expectation Maximization (EM) Colin Dewey February 14, 2008 1 Sequence Motifs what is a sequence motif? a sequence pattern of biological significance typically
Codon Distribution in Error-Detecting Circular Codes
 life Article Codon Distribution in Error-Detecting Circular Codes Elena Fimmel, * and Lutz Strüngmann Institute for Mathematical Biology, Faculty of Computer Science, Mannheim University of Applied Sciences,
life Article Codon Distribution in Error-Detecting Circular Codes Elena Fimmel, * and Lutz Strüngmann Institute for Mathematical Biology, Faculty of Computer Science, Mannheim University of Applied Sciences,
Searching Sear ( Sub- (Sub )Strings Ulf Leser
Strings Ulf Leser") Searching (Sub-)Strings Ulf Leser This Lecture Exact substring search Naïve Boyer-Moore Searching with profiles Sequence profiles Ungapped approximate search Statistical evaluation of search results Ulf
Searching (Sub-)Strings Ulf Leser This Lecture Exact substring search Naïve Boyer-Moore Searching with profiles Sequence profiles Ungapped approximate search Statistical evaluation of search results Ulf
Data Mining in Bioinformatics HMM
 Data Mining in Bioinformatics HMM Microarray Problem: Major Objective n Major Objective: Discover a comprehensive theory of life s organization at the molecular level 2 1 Data Mining in Bioinformatics
Data Mining in Bioinformatics HMM Microarray Problem: Major Objective n Major Objective: Discover a comprehensive theory of life s organization at the molecular level 2 1 Data Mining in Bioinformatics
The Trigram and other Fundamental Philosophies
 The Trigram and other Fundamental Philosophies by Weimin Kwauk July 2012 The following offers a minimal introduction to the trigram and other Chinese fundamental philosophies. A trigram consists of three
The Trigram and other Fundamental Philosophies by Weimin Kwauk July 2012 The following offers a minimal introduction to the trigram and other Chinese fundamental philosophies. A trigram consists of three
Lecture IV A. Shannon s theory of noisy channels and molecular codes
 Lecture IV A Shannon s theory of noisy channels and molecular codes Noisy molecular codes: Rate-Distortion theory S Mapping M Channel/Code = mapping between two molecular spaces. Two functionals determine
Lecture IV A Shannon s theory of noisy channels and molecular codes Noisy molecular codes: Rate-Distortion theory S Mapping M Channel/Code = mapping between two molecular spaces. Two functionals determine
The role of the FliD C-terminal domain in pentamer formation and
 The role of the FliD C-terminal domain in pentamer formation and interaction with FliT Hee Jung Kim 1,2,*, Woongjae Yoo 3,*, Kyeong Sik Jin 4, Sangryeol Ryu 3,5 & Hyung Ho Lee 1, 1 Department of Chemistry,
The role of the FliD C-terminal domain in pentamer formation and interaction with FliT Hee Jung Kim 1,2,*, Woongjae Yoo 3,*, Kyeong Sik Jin 4, Sangryeol Ryu 3,5 & Hyung Ho Lee 1, 1 Department of Chemistry,
part 4: phenomenological load and biological inference. phenomenological load review types of models. Gαβ = 8π Tαβ. Newton.
 2017-07-29 part 4: and biological inference review types of models phenomenological Newton F= Gm1m2 r2 mechanistic Einstein Gαβ = 8π Tαβ 1 molecular evolution is process and pattern process pattern MutSel
2017-07-29 part 4: and biological inference review types of models phenomenological Newton F= Gm1m2 r2 mechanistic Einstein Gαβ = 8π Tαβ 1 molecular evolution is process and pattern process pattern MutSel
Evolutionary Analysis of Viral Genomes
 University of Oxford, Department of Zoology Evolutionary Biology Group Department of Zoology University of Oxford South Parks Road Oxford OX1 3PS, U.K. Fax: +44 1865 271249 Evolutionary Analysis of Viral
University of Oxford, Department of Zoology Evolutionary Biology Group Department of Zoology University of Oxford South Parks Road Oxford OX1 3PS, U.K. Fax: +44 1865 271249 Evolutionary Analysis of Viral
Quantitative Bioinformatics
 Chapter 9 Class Notes Signals in DNA 9.1. The Biological Problem: since proteins cannot read, how do they recognize nucleotides such as A, C, G, T? Although only approximate, proteins actually recognize
Chapter 9 Class Notes Signals in DNA 9.1. The Biological Problem: since proteins cannot read, how do they recognize nucleotides such as A, C, G, T? Although only approximate, proteins actually recognize
Casting Polymer Nets To Optimize Molecular Codes
 Casting Polymer Nets To Optimize Molecular Codes The physical language of molecules Mathematical Biology Forum (PRL 2007, PNAS 2008, Phys Bio 2008, J Theo Bio 2007, E J Lin Alg 2007) Biological information
Casting Polymer Nets To Optimize Molecular Codes The physical language of molecules Mathematical Biology Forum (PRL 2007, PNAS 2008, Phys Bio 2008, J Theo Bio 2007, E J Lin Alg 2007) Biological information
Exact Algorithms for Planted Motif Problems CONTACT AUTHOR:
 Exact Algorithms for Planted Motif Problems CONTACT AUTHOR: Sanguthevar Rajasekaran 257 ITE Building, Dept. of CSE Univ. of Connecticut, Storrs, CT 06269-2155 rajasek@engr.uconn.edu (860) 486 2428; (860)
Exact Algorithms for Planted Motif Problems CONTACT AUTHOR: Sanguthevar Rajasekaran 257 ITE Building, Dept. of CSE Univ. of Connecticut, Storrs, CT 06269-2155 rajasek@engr.uconn.edu (860) 486 2428; (860)
Motifs and Logos. Six Introduction to Bioinformatics. Importance and Abundance of Motifs. Getting the CDS. From DNA to Protein 6.1.
 Motifs and Logos Six Discovering Genomics, Proteomics, and Bioinformatics by A. Malcolm Campbell and Laurie J. Heyer Chapter 2 Genome Sequence Acquisition and Analysis Sami Khuri Department of Computer
Motifs and Logos Six Discovering Genomics, Proteomics, and Bioinformatics by A. Malcolm Campbell and Laurie J. Heyer Chapter 2 Genome Sequence Acquisition and Analysis Sami Khuri Department of Computer
CSCI 4181 / CSCI 6802 Algorithms in Bioinformatics
 CSCI 4181 / CSCI 6802 Algorithms in Bioinformatics 1 "In science there is only physics; all the rest is stamp collecting." -Ernest Rutherford 2 Was I a stamp collector? Alan Turing 3 Inte skert, men vad
CSCI 4181 / CSCI 6802 Algorithms in Bioinformatics 1 "In science there is only physics; all the rest is stamp collecting." -Ernest Rutherford 2 Was I a stamp collector? Alan Turing 3 Inte skert, men vad
Neyman-Pearson. More Motifs. Weight Matrix Models. What s best WMM?
 Neyman-Pearson More Motifs WMM, log odds scores, Neyman-Pearson, background; Greedy & EM for motif discovery Given a sample x 1, x 2,..., x n, from a distribution f(... #) with parameter #, want to test
Neyman-Pearson More Motifs WMM, log odds scores, Neyman-Pearson, background; Greedy & EM for motif discovery Given a sample x 1, x 2,..., x n, from a distribution f(... #) with parameter #, want to test
Sex-Linked Inheritance in Macaque Monkeys: Implications for Effective Population Size and Dispersal to Sulawesi
 Supporting Information http://www.genetics.org/cgi/content/full/genetics.110.116228/dc1 Sex-Linked Inheritance in Macaque Monkeys: Implications for Effective Population Size and Dispersal to Sulawesi Ben
Supporting Information http://www.genetics.org/cgi/content/full/genetics.110.116228/dc1 Sex-Linked Inheritance in Macaque Monkeys: Implications for Effective Population Size and Dispersal to Sulawesi Ben
AtTIL-P91V. AtTIL-P92V. AtTIL-P95V. AtTIL-P98V YFP-HPR
 Online Resource 1. Primers used to generate constructs AtTIL-P91V, AtTIL-P92V, AtTIL-P95V and AtTIL-P98V and YFP(HPR) using overlapping PCR. pentr/d- TOPO-AtTIL was used as template to generate the constructs
Online Resource 1. Primers used to generate constructs AtTIL-P91V, AtTIL-P92V, AtTIL-P95V and AtTIL-P98V and YFP(HPR) using overlapping PCR. pentr/d- TOPO-AtTIL was used as template to generate the constructs
"Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky
 MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
Greedy Algorithms. CS 498 SS Saurabh Sinha
 Greedy Algorithms CS 498 SS Saurabh Sinha Chapter 5.5 A greedy approach to the motif finding problem Given t sequences of length n each, to find a profile matrix of length l. Enumerative approach O(l n
Greedy Algorithms CS 498 SS Saurabh Sinha Chapter 5.5 A greedy approach to the motif finding problem Given t sequences of length n each, to find a profile matrix of length l. Enumerative approach O(l n
Plan. Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics.
 and its used in Chemoinformatics.") Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Re- engineering cellular physiology by rewiring high- level global regulatory genes
 Re- engineering cellular physiology by rewiring high- level global regulatory genes Stephen Fitzgerald 1,2,, Shane C Dillon 1, Tzu- Chiao Chao 2, Heather L Wiencko 3, Karsten Hokamp 3, Andrew DS Cameron
Re- engineering cellular physiology by rewiring high- level global regulatory genes Stephen Fitzgerald 1,2,, Shane C Dillon 1, Tzu- Chiao Chao 2, Heather L Wiencko 3, Karsten Hokamp 3, Andrew DS Cameron
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment
 Pairwise sequence alignment") Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Computational Genomics. Systems biology. Putting it together: Data integration using graphical models
 02-710 Computational Genomics Systems biology Putting it together: Data integration using graphical models High throughput data So far in this class we discussed several different types of high throughput
02-710 Computational Genomics Systems biology Putting it together: Data integration using graphical models High throughput data So far in this class we discussed several different types of high throughput
Estimating Phred scores of Illumina base calls by logistic regression and sparse modeling
 Zhang et al. BMC Bioinformatics (2017) 18:335 DOI 10.1186/s12859-017-1743-4 RESEARCH ARTICLE Open Access Estimating Phred scores of Illumina base calls by logistic regression and sparse modeling Sheng
Zhang et al. BMC Bioinformatics (2017) 18:335 DOI 10.1186/s12859-017-1743-4 RESEARCH ARTICLE Open Access Estimating Phred scores of Illumina base calls by logistic regression and sparse modeling Sheng
BIOINFORMATICS. Neighbourhood Thresholding for Projection-Based Motif Discovery. James King, Warren Cheung and Holger H. Hoos
 BIOINFORMATICS Vol. no. 25 Pages 7 Neighbourhood Thresholding for Projection-Based Motif Discovery James King, Warren Cheung and Holger H. Hoos University of British Columbia Department of Computer Science
BIOINFORMATICS Vol. no. 25 Pages 7 Neighbourhood Thresholding for Projection-Based Motif Discovery James King, Warren Cheung and Holger H. Hoos University of British Columbia Department of Computer Science
In previous lecture. Shannon s information measure x. Intuitive notion: H = number of required yes/no questions.
 In previous lecture Shannon s information measure H ( X ) p log p log p x x 2 x 2 x Intuitive notion: H = number of required yes/no questions. The basic information unit is bit = 1 yes/no question or coin
In previous lecture Shannon s information measure H ( X ) p log p log p x x 2 x 2 x Intuitive notion: H = number of required yes/no questions. The basic information unit is bit = 1 yes/no question or coin
CSE 417T: Introduction to Machine Learning. Final Review. Henry Chai 12/4/18
 CSE 417T: Introduction to Machine Learning Final Review Henry Chai 12/4/18 Overfitting Overfitting is fitting the training data more than is warranted Fitting noise rather than signal 2 Estimating! "#$
CSE 417T: Introduction to Machine Learning Final Review Henry Chai 12/4/18 Overfitting Overfitting is fitting the training data more than is warranted Fitting noise rather than signal 2 Estimating! "#$
Timing molecular motion and production with a synthetic transcriptional clock
 Timing molecular motion and production with a synthetic transcriptional clock Elisa Franco,1, Eike Friedrichs 2, Jongmin Kim 3, Ralf Jungmann 2, Richard Murray 1, Erik Winfree 3,4,5, and Friedrich C. Simmel
Timing molecular motion and production with a synthetic transcriptional clock Elisa Franco,1, Eike Friedrichs 2, Jongmin Kim 3, Ralf Jungmann 2, Richard Murray 1, Erik Winfree 3,4,5, and Friedrich C. Simmel
Predicting Protein Functions and Domain Interactions from Protein Interactions
 Predicting Protein Functions and Domain Interactions from Protein Interactions Fengzhu Sun, PhD Center for Computational and Experimental Genomics University of Southern California Outline High-throughput
Predicting Protein Functions and Domain Interactions from Protein Interactions Fengzhu Sun, PhD Center for Computational and Experimental Genomics University of Southern California Outline High-throughput
Matrix-based pattern discovery algorithms
 Regulatory Sequence Analysis Matrix-based pattern discovery algorithms Jacques.van.Helden@ulb.ac.be Université Libre de Bruxelles, Belgique Laboratoire de Bioinformatique des Génomes et des Réseaux (BiGRe)
Regulatory Sequence Analysis Matrix-based pattern discovery algorithms Jacques.van.Helden@ulb.ac.be Université Libre de Bruxelles, Belgique Laboratoire de Bioinformatique des Génomes et des Réseaux (BiGRe)
ChemiScreen CaS Calcium Sensor Receptor Stable Cell Line
 PRODUCT DATASHEET ChemiScreen CaS Calcium Sensor Receptor Stable Cell Line CATALOG NUMBER: HTS137C CONTENTS: 2 vials of mycoplasma-free cells, 1 ml per vial. STORAGE: Vials are to be stored in liquid N
PRODUCT DATASHEET ChemiScreen CaS Calcium Sensor Receptor Stable Cell Line CATALOG NUMBER: HTS137C CONTENTS: 2 vials of mycoplasma-free cells, 1 ml per vial. STORAGE: Vials are to be stored in liquid N
Bioinformatics Chapter 1. Introduction
 Bioinformatics Chapter 1. Introduction Outline! Biological Data in Digital Symbol Sequences! Genomes Diversity, Size, and Structure! Proteins and Proteomes! On the Information Content of Biological Sequences!
Bioinformatics Chapter 1. Introduction Outline! Biological Data in Digital Symbol Sequences! Genomes Diversity, Size, and Structure! Proteins and Proteomes! On the Information Content of Biological Sequences!
Model Accuracy Measures
 Model Accuracy Measures Master in Bioinformatics UPF 2017-2018 Eduardo Eyras Computational Genomics Pompeu Fabra University - ICREA Barcelona, Spain Variables What we can measure (attributes) Hypotheses
Model Accuracy Measures Master in Bioinformatics UPF 2017-2018 Eduardo Eyras Computational Genomics Pompeu Fabra University - ICREA Barcelona, Spain Variables What we can measure (attributes) Hypotheses
BLAST: Basic Local Alignment Search Tool
 .. CSC 448 Bioinformatics Algorithms Alexander Dekhtyar.. (Rapid) Local Sequence Alignment BLAST BLAST: Basic Local Alignment Search Tool BLAST is a family of rapid approximate local alignment algorithms[2].
.. CSC 448 Bioinformatics Algorithms Alexander Dekhtyar.. (Rapid) Local Sequence Alignment BLAST BLAST: Basic Local Alignment Search Tool BLAST is a family of rapid approximate local alignment algorithms[2].
6.047 / Computational Biology: Genomes, Networks, Evolution Fall 2008
 MIT OpenCourseWare http://ocw.mit.edu 6.047 / 6.878 Computational Biology: Genomes, etworks, Evolution Fall 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms.
MIT OpenCourseWare http://ocw.mit.edu 6.047 / 6.878 Computational Biology: Genomes, etworks, Evolution Fall 2008 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms.
10-810: Advanced Algorithms and Models for Computational Biology. microrna and Whole Genome Comparison
 10-810: Advanced Algorithms and Models for Computational Biology microrna and Whole Genome Comparison Central Dogma: 90s Transcription factors DNA transcription mrna translation Proteins Central Dogma:
10-810: Advanced Algorithms and Models for Computational Biology microrna and Whole Genome Comparison Central Dogma: 90s Transcription factors DNA transcription mrna translation Proteins Central Dogma:
Graph Alignment and Biological Networks
 Graph Alignment and Biological Networks Johannes Berg http://www.uni-koeln.de/ berg Institute for Theoretical Physics University of Cologne Germany p.1/12 Networks in molecular biology New large-scale
Graph Alignment and Biological Networks Johannes Berg http://www.uni-koeln.de/ berg Institute for Theoretical Physics University of Cologne Germany p.1/12 Networks in molecular biology New large-scale
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Insects act as vectors for a number of important diseases of
 pubs.acs.org/synthbio Novel Synthetic Medea Selfish Genetic Elements Drive Population Replacement in Drosophila; a Theoretical Exploration of Medea- Dependent Population Suppression Omar S. Abari,,# Chun-Hong
pubs.acs.org/synthbio Novel Synthetic Medea Selfish Genetic Elements Drive Population Replacement in Drosophila; a Theoretical Exploration of Medea- Dependent Population Suppression Omar S. Abari,,# Chun-Hong
Support Vector Machine & Its Applications
 Support Vector Machine & Its Applications A portion (1/3) of the slides are taken from Prof. Andrew Moore s SVM tutorial at http://www.cs.cmu.edu/~awm/tutorials Mingyue Tan The University of British Columbia
Support Vector Machine & Its Applications A portion (1/3) of the slides are taken from Prof. Andrew Moore s SVM tutorial at http://www.cs.cmu.edu/~awm/tutorials Mingyue Tan The University of British Columbia