G4120: Introduction to Computational Biology
|
|
|
- Valentine Fox
- 6 years ago
- Views:
Transcription
1 ICB Fall 2003 G4120: Introduction to Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2003 Oliver Jovanovic, All Rights Reserved.
2 Bioinformatics and Computational Biology Internet Resources National Center for Biotechnology Information (NCBI) PubMed, PubMed Central, Books and other reference material GenBank, RefSeq, CDD, MMDB and other sequence and structure databases Prokaryotic genome data and browsers (over 100 microbial, 1,000 virus and 300 plasmids) Eukaryotic genome data and browsers (9 complete genomes, maps and partial sequences) BLAST, PSI-BLAST and VAST search tools, Cn3D visualization tool Ensembl (EMBL-EBI/Sanger Institute) Eukaryotic genome data and browsers (human, mouse, rat,fugu, zebrafish, mosquito, Drosophila, C. elegans, and C. briggsae). UCSC Genome Bioninformatics Eukaryotic genome data and browsers (human, mouse, rat). European Bioinformatics Institute Sequence analysis tools and databases Expert Protein Analysis System (Expasy) Protein analysis and biochemical information, links to useful tools, software and references. Protein Data Bank Worldwide repository for 3D protein structure data and tools.
3 Bioinformatics and Computational Biology Software Resources IU Bio-Archive (Macintosh, Unix and Java Molecular Biology Software) Pasteur Institute Macintosh Bioinformatics Archive ftp://ftp.pasteur.fr/pub/gensoft/macintosh/ European Bioinformatics Institute Biology Software Directory Apple Computer Bioinformatics Ports to Mac OS X European Molecular Biology Open Software Suite (EMBOSS) BioTeam, Inc. Bioinformatics Tools Ports to Mac OS X Fink Scientific Tool Ports to Mac OS X SourceForge VersionTracker
4 Databases Flat File Database (FFDB) A collection of similar files made useful by ordering and indexing. All the information about one sequence would be stored in one structured text file, and you generally examine one file at a time. Examples: GenBank, FileMaker Pro Relational Database (RDB) All data is stored inside one or more tables of rows and column, with all operations done on the tables themselves or producing other tables as the result. All the information about one sequence would be stored in a collection of tables with other data, so you can easily look at just the information relating to that sequence, or how it relates to the database as a whole. Structured Query Language (SQL) is used to access data in a relational database. Examples: msql, MySQL, PostgreSQL, Microsoft SQL Server, Oracle Object Oriented Databases (OODB) Data is stored and retrieved in an fashion consistent with object oriented programming principles (based on languages such as Smalltalk, C++ or Java). They generally handle complex structures and concurrent interaction by multiple clients well. Many relational databases have or are acquiring object oriented database features. Examples: PDB, Versant VDB, Gemstone GemFire
5 Searching Sequence Databases Needleman-Wunsch Needleman-Wunsch gives you the optimal global alignment of two sequences. This is best for comparing closely related sequences of similar lengths. Examples: GCG Gap, EMBOSS Needle Smith-Waterman Smith-Waterman gives you the optimal local alignment of two sequences. This is better for comparing distantly related sequences (where non-functional regions may have diverged). Examples: GCG BestFit, EMBOSS Water BLAST BLAST gives a fast approximation of Smith-Waterman, from 100-1,000 times faster, but will not necessarily find optimal local alignments. Examples: NCBI BLAST, WU-BLAST
6 Rules of Thumb for BLAST The shortest possible word size (2 for proteins, 7 for nucleotides) gives the most sensitivity, though the search may take more time. Note: A larger word size (3 for proteins, 11 for nucleotides) is the default setting for NCBI BLAST. You will have to change it manually. At least initially, run your search with the Low Complexity filter off. Then, if you appear to be getting spurious hits, or for comparison purpose, run it again with the filter on. Although it can be helpful, the filter can also filter out a significant match. Note: Filter on the default setting for NCBI BLAST. You will have to turn it off manually. Keep in mind that BLAST is a heuristic version of Smith-Waterman, and may miss a significant alignment. The default BLOSUM 62 substitution scoring matrix is best for comparing moderately distant and relatively closely related proteins. When searching for distantly related proteins, try the PAM 250 and BLOSUM 45 matrices. If comparing closely related proteins, try the PAM 1 and BLOSUM 80 matrices. PSI-BLAST can be useful for searching for very weak protein homologies. If searching with short DNA or protein sequences make sure you use the appropriate Search for short nearly exact matches BLAST page, or make sure to use those settings. BLAST is not the best tool to use for very short sequences. The Limit by entrez query option allows BLAST searches to be limited to the results of an Entrez query against the database chosen, typically one or more organisms. Common organisms are provided in a popup menu. This can yield more relevant results.
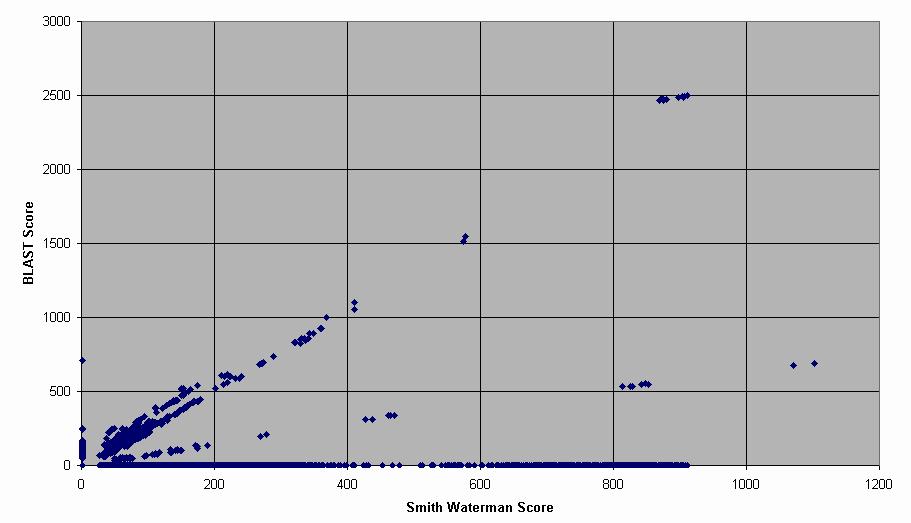
7 BLAST vs. Smith-Waterman

8 Blosum 62 vs. PAM 250
9 Rules of Thumb for Significance of Protein Alignments Protein Identity Significance Under 20% Unlikely to be significant 20% to 30% Gray zone may or may not be significant Over 30% Likely to be significant Keep in mind that when searching GenBank with a protein sequence it is possible to get results with a stretch of amino acids with over 50% identity by chance alone. Identity throughout an entire protein is more likely to be significant, however, homologous proteins with a very low level of identity exist. Such distant relatives can be identified through comparison to other homologous proteins. Identity within known functional domains is more likely to be significant, and may suggest functional homology.
10 Definitions Identity - the extent to which two sequences are invariant. Similarity - the extent to which sequences are related, based on sequence identity and/or conservation. Conservation - changes in an amino acid sequence that preserve the biochemical properties of the original residue. This is measured in most sequence comparison algorithms by substitution matrices in which scores for each position are derived from observations of the frequencies of substitutions in blocks of local alignments in related proteins. Homology - similarity attributed to descent from a common ancestor. It may or may not result in similar function. Orthologous - homologous sequences in different species that arose from a common ancestral gene. Paralogous - homologous sequences within a single species that arose by gene duplication.
11 Multiple Multiple (MSA) A multiple sequence alignment is an alignment of a set of sequences with structurally similar and evolutionarily homologous residues aligned in columns. In an ideal alignment, columns of aligned amino acid residues would have similar locations in the 3D structure of a protein and would diverge from a common ancestral residue. In theory, an unambigously correct evolutionary alignment exists, but can be difficult to infer and computationally intensive to calculate. Where structural data is lacking or limited, as is generally the case, it is not possible to unambiguously identify structurally similar positions. Thus, defining a single unambiguous ideal alignment can be very difficult.
12 Multiple Algorithms Dynamic Programming vs. Heuristic Alignment Using dynamic programming algorithms (such as Smith-Waterman or Needleman-Wunsch) to perform an optimal alignment of more than a few sequences is computationally intensive, and generally impractical for large sets of sequences or lengthy sequences. As a result, most commonly used multiple sequence alignment algorithms take a heuristic approach. One common heuristic approach is progressive alignment, in which the problem is broken down into a series of pairwise alignments. The details of how to choose the initial pair to align, how to score alignments, how to align subsequent sequences, and whether subfamilies of alignments should be created can all vary. MSA (Dynamic) This algorithm uses a technique that reduces the complexity of dynamic programming when applied to multiple sequences, and can give an optimal alignment for seven short ( aa) protein sequences in a reasonable amount of time. For alignments with more or longer sequences, a heuristic approach is more practical. Feng-Doolittle (Heuristic) One of the first progressive alignment algorithms. It does not take advantage of profiles, which can increase the accuracy of the alignment. ClustalW (Heuristic) This profile based progressive alignment algorithm uses a number of heuristics to generate multiple sequence alignments, including phylogeny and scalable gap penalties.
13 Multiple with Text Use Fixed Width or Monospaced Fonts Each character in the font takes up the same amount of horizontal space, allowing multiple sequence alignments to properly align. Examples: Andale Mono, Courier, Courier New, Monaco, V100 Fixed Width Font Alignment (Courier):... m s h N q f q f i G n L t r D M A s R G v N K V I L V G n L G q D M A v R G I N K V I L V G R L G k D Variable Width Font Alignment (Times):... m s h N q f q f i G n L t r D M A s R G v N K V I L V G n L G q D M A v R G I N K V I L V G R L G k D
14 Multiple with Excel 1 50 RK2... m s h N q f q f i G n L t r D t E V R h g n s n k p q A i f d i A v n E e W R n d a. G d k E. coli M A s R G v N K V I L V G n L G q D P E V R Y m P N G G A V A N i t l A T S E S W R D K a T G E M F M A v R G I N K V I L V G R L G k D P E V R Y I P N G G A V A N L Q V A T S E S W R D K Q T G E M ColIb-P9 M s a R G I N K V I L V G R L G n D P E V R Y I P N G G A V A N L Q V A T S E S W R D K Q T G E M R64 M s a R G I N K V I L V G R L G n D P E V R Y I P N G G A V A N L Q V A T S E S W R D K Q T G E M pip71a M A v R G I N K V I L V G R L G k D P E V R Y I P N G G A V A N L Q V A T S E S W R D K Q T G E i pip231a M A v R G I N K V I L V G R L G k D P E V R Y I P N G G A V A N L Q V A T S E t W R D K Q T G K M RK2 q E r T d f f R i k c F G s q A E a h G k Y L g K G s l V f v q G k i R n t k y E k d. G q T v Y E. coli k E Q T E W H R V V L F G K L A E V A s E Y L R K G s Q V Y I E G Q L R T R k W t D q s G q d R Y F R E Q T E W H R V V L F G K L A E V A G E c L R K G A Q V Y I E G Q L R T R S W E D N. G I T R Y ColIb-P9 R E Q T E W H R V V L F G K L A E V A G E Y L R K G A Q V Y I E G Q L R T R S W d D N. G I T R Y R64 R E Q T E W H R V V L F G K L A E V A G E Y L R K G A Q V Y I E G Q L R T R S W d D N. G I T R Y pip71a R E Q T E W H R V V L F G K L A E V A G E Y L R K G A Q V Y I E G Q L R T R S W E D N. G I T R Y pip231a R E Q T E W H R V V L F G K L A E V A G E Y L R K G A Q V Y I E G Q L R T R S W E D N. G I T R Y RK2 g T d f.. i a d k v d y l d t k A p G g s n Q e E. coli t T E v v V n v g G T M Q M L G g r q G g g a p a g g n i g g. G Q P Q s g w g q p q q p q g G n F v T E I L V K T T G T M Q M L v r A a G a q t Q p e e g q Q f s G Q P Q p e p q a E a g t K K G G ColIb-P9 i T E I L V K T T G T M Q M L G s A p q q n a Q a q p k p Q q n G Q P Q s a d a t.... K K G G R64 i T E I L V K T T G T M Q M L G s A p q q n a Q a q p k p Q q n G Q P Q s a d a t.... K K G G pip71a v T E I L V K T T G T M Q M L G r A a G t q t Q p e e a q Q f s G Q P Q p e s q p E p.. K K G G pip231a v T E I L V K T T G T M Q M L G r A a G a q t Q p e e g q Q s a. Q P Q p e p q s E a g t K K G G % Identity % Similarity RK E. coli q f s g G a q s r p q Q s a P a a p s n E p p m d f d. D D I P F F A K T K G R g R K A A Q P E P Q p Q p P E G d D Y G F S D D I P F ColIb-P9 A K T K G R g R K A A Q P E P Q p Q t P E G e D Y G F S D D I P F R64 A K T K G R e R K A A Q P E P Q p Q t P E G e D Y G F S D D I P F pip71a A K T K G R e R K A A Q P E P r q p s e p a.. Y D F d D D I P F pip231a A K T K G R g R K V A Q P E P Q l Q p P E G d D Y G F S D D I P F Can use with any font, as Excel allows you to manually adjust the alignment.
15 ClustalW and ClustalX ClustalW ClustalW first generates a pairwise distance matrix for all the sequences by pairwise dynamic programming alignment. It then estimates evolutionary distance from similarity scores and constructs a guide tree using the neighbor joining distance matrix method. Dynamic progamming is then used to align the most closely related pairs of sequences. A sequence profile is constructed from these alignments, and the remaining sequences are progressively aligned to each other in order of decreasing similarity by profile-profile, profile-sequence or sequence-sequence alignment, until a complete multiple sequence alignment has been generated. ClustalW automatically chooses the optimal scoring matrix for protein alignments based on whether the sequences are close or distant neighbors in the tree. Thus it might use BLOSUM 62 (optimal for close relationships) for close neighbors, and BLOSUM45 (optimal for distant neighbors) for distant neighbors. ClustalW also allows for scalable gap penalties in protein profile alignments. A gap opening next to a highly conserved residue can be more heavily penalized than a gap opening next to an unconserved residue, for example. ClustalX This is a version of ClustalW with a graphical user interface, which is more intuitive to use, though the formatting requirements for input files need to be followed closely. It can display multiple sequence alignments onscreen, or output them as Postscript, which can easily be converted to PDF format by ESP GhostScript with GStill.
16 Multiple with ClustalX CLUSTAL X (1.82) MULTIPLE SEQUENCE ALIGNMENT File: tadafasta.ps Date: Wed Apr 2 12:19: Page 1 of 2 ::.. : * :. : ::: * **:. *: V_fisch MDQNKSIYIEIRAQIFDVLD--AETVN SLSKE--QLHNQLSN AIDLLIERHEWPVSTIVRAEYVTSLVNELQGLGPLQVLM 77 V_fisch MNNNKALYIQLRTQIFNALE--PEALN KLTKQ--ELTQQLSN AVDLLIDREQLPVSLIMKNEYVESLVNELVGLGPLQNLM 77 V_vulnII1_ MNQLKQIYLDLRDEIFDAID--ASTLS EISNE--ELAEQLSE SVNILIDKKQLQVSSLKRAELVKALYDELKGLGPLQKLV 77 Y_pes MIVPLKIQELMRERMLANID--INKVE LLVGDRNKLIGLLSQ TFDDLFNNNEYNLTTQAQKYIIEMIADEITGFGPLRELM 79 Y_ent MLASID--IDQVQ YLVDDYSKLSELLSQ TLDELFNNNDYKLTTQDQKKIITMIADEITGFGPLRELM 65 A_act MLTKQQKILLRSEVLSNLD--IEKID ELQSERSSLVNELVQ IVNRVANKSGAYLTSADTLVMAEIVADEIEGYGPLRDLM 78 H_aph MLTKEQQIFLRSEVLSNLD--IEKID ALQSERNLLVNELVQ IVNRVASKSGTYLTSADTLVMAEIVADEIEGYGPLRDLM 78 P_mul MLTKEQQVFFRNELLSNLD--IEKID EIQSERDKLVDELVQ VVYKVAGKGNIYITSADALFMAECIADEIDGYGPIRELM 78 H_duc MLTKDQQVFFRNALLSNLN--VDTLD EIENERSKLVTELTQ SLYRVANTNNIYITPYDATDMAEIVADEIGGYGPIRELM 78 A_pleur MLTKEQQIFFRTELLSNLD--VEKLD EIQNERNKLIDELTQ SLYRISNLHSIYLTPADAAYMAGLVADEIGGYGPIRELM 78 V_vulnI8_11 MFGN KTQMVNVSRGNPLVMPEAAQTAFEKLIEPSE AVKLTRKQLQQEIKK AVAQLSAQ-QLLPYNQSELAILVEQLCDDMLGVGPIQCLV 89 V_vulnI6_11 MFFKRKNINPEFQEKAAALEAQPSSTISDEVISDIESNVQPIDSNRVEPMQQDKKLLERQAKDKAVEEARKQLEQELAIKHYYHQRLLETLDLGLLSSLEKERAKKDLHDAIVQLMAEDQTHPMSSEGRKRVIKQIEDEVFGLGPLEPLL 150 ruler : :.**::** :::* * : *.. :* :.*:..**:*:. * *:** ****:* *:*::*. :***:*. : :.::: : :.. **:::****:**** ** :* * :*:: V_fisch1 EDESISDIMINGYDKIFIERAGLVEVAPVSFIDEEQLLHIAKRVASQVGRRVDDSSPTCDARLADGSRVNIVIPPIAIDGTSMSIRKFKKDSIGLEKLTEFGALSQEMAQLLMIASRCRLNILISGGTGSGKTTMLNALSQYISEKERIV 227 V_fisch2 DDETITDIMINGHENVFIERDGLVEKVSVNFIDEQQLIDIAKRIASRVGRRVDESSPTCDARLEDGSRVNIVIPPIAIDGTSISIRKFKKQSIAFSDLVEFGAMSKEMAQILMVASRCRLNILISGGTGSGKTTMLNALSQFISEGERIV 227 V_vulnII1_6 ENDDISDIMINGPYDVFIEIGGKVEKSPIQFVNEKQLNTIAKRIASNVGRRIDESSPLCDARLKDGSRVNIVIPPLAIDGTSISIRKFKEQKIKLENLVEFGAMSIEMAKLLSIASHCKCNILISGGTGSGKTTLLNALSGFIGEGERVV 227 Y_pes EDDSISDIMVNGPERIFIERYGLLKLTDRRFVNNTQLTDIAKRLMQKVNRRIDEGRPLADARLIDGSRINVAISPIALDGTALSIRKFSKNKRRLEDLVDMGAMSSDMANFLIIAASCRVNIIISGGTGSGKTTLLNALSKYISEDERVI 229 Y_ent EDDSISDIMVNGPEKIFIERFGMITLTSRRFINNAQLTDIAKRLMQRANRRIDEGRPLADARLIDGSRINVAISPIALDGTVLSIRKFSNNKRKLEDLVEMGAMSSDMANFLIIAASCRVNIIISGGTGSGKTTLLNALSMYISENERVI 215 A_act ADDTINDILVNGPNDIWVERAGILEKTDKEFVSNEQLTDIAKRLVARVGRRIDDGSPLVDSRLPDGSRLNAVIAPIALDGTSISIRKFSKNKKTLQELVNFGSMTRNGE-FLNYCCRSRVNIIVSGGTGSGKTTLLNALSNYISHTERVI 227 H_aph ADDTINDILVNGPDDVWIERAGILEKTSKEFVSNEQLTDIAKRLVARVGRRIDDGSPLVDSRLPDGSRLNVVIAPIALDGTSVSIRKFSKNKKTLQELVNFGSMTREMANFLIIAARSRVNIIVSGGTGSGKTTLLNALSNYISHSERVI 228 P_mul EDETVNDILVNGPDDVWVERAGILEKTDKKFISNEQLTDIAKRLVAKVGRRIDDGSPLVDSRLPDGSRLNVVIAPIALDGTSISIRKFSKSKKSLQELVNFGSMTREMANFLIIAARSRVNIIVSGGTGSGKTTLLNALSNYISPKERVI 228 H_duc EDDTVNDILVNGPDNIWIERAGVLEKTNKTFINNEQLTDIAKRLVARVGRRIDEGMPLVDSRLPDGSRLNVVIQPIALDGTSISIRKFSKSKKSLQELVNFGSMTLDMANFLIIAARSRVNIIVSGGTGSGKTTLLNALSSYISPTERVL 228 A_pleur EDEGVNDILVNGPDNIWVERAGILEKTDKKFINNEQLTDIAKRLVARVGRRIDEGMPLVDSRLPDGSRLNVVIQPIALDGTSISIRKFSKSKKSLQDLVNYGSMTLDMANFLIIAARSRVNIIVSGGTGSGKTTLLNALSHYISHTERVL 228 V_vulnI8_11 EDPSVSDILVNGPEQIYIERQGKLLKTDIRFRDKKHLLNVAQRIVNAVGRRLDESTPLVDARLEDGSRVNIIAPPLALNGVCISIRKFPERQYDLPGLVAFGSLSEEMAQCLALAARCRLNILVSGGTGAGKTTLLNAMSTPISDDERII 239 V_vulnI6_11 HDKTVSDILVNGPKNIFVERRGKLEKTPYTFLDDRHLRNIIDRIVSQVGRRIDEASPMVDARLLDGSRVNAIIPPLALDGASVSIRRFAVDKLTMDNMLGYNSLSPQMAKFVEAAVKGELNILIAGGTGSGKTTTLNIFSGFIPSDDRII 300 ruler *:**:*** * :** :::***.. *.* :: :*** *:*****:**::** ** *:.:** ******:**:.*:***:* ** * * *. *. :. :* * **:.:::* *.**.*:: * : *:* : : :: V_fisch1 TIEDAAELKLLQPHVVRLETRNSGIEGNGAITQQDLVINALRMRPDRIIVGECRGGEAFQMLQAMNTGHDGSMSTLHANTPRDAMARVEAMVMMASNNLPLEAIRRTIVSAVDIVIQISRLHDGSRKVMSITEVIGLEGNNVVLEELYKF 377 V_fisch2 TIEDAAELKLQQPHVVRLETRTSGIEGTGVVSQRDLVINSLRMRPDRIIVGECRGGEAFEMLQAMNTGHDGSMSTLHANSPRDALSRVEAMVMMATNNLPLEAVRRTIVSAVDIVIQISRLHDGTRKVMSISEVVGLEGNNVVLEEIFAF 377 V_vulnII1_6 TIEDAAELQLQKPHIVRLETRQASVEGTGQITARDLVINALRMRPDRIIVGECRGAEAFEMLQAMNTGHDGSMSTLHANTPRDAIARTESMVMMATASLPLEAIRRTIVSAVDLIVQVRRLHDGSRKVMYISEIVGLEGNNVVMEDIFRF 377 Y_pes TLEDAAELNLEQPHVVRMETRLAGLENTGQITMRDLVINSLRMRPDRIIIGECRGEETFEMLQAMNTGHNGSMSTLHANTPRDAVARLESMIMMGPVNMPLITIRRNIASAINLIVQVSRMNDGSRKIRNISEIMGMEGEHVVLQDIFTF 379 Y_ent TLEDAAELNLEQPHVVRMETRLAGLENTGQITMRDLVINSLRMRPDRIIIGECRGEETFEMLQAMNTGHNGSMSTLHANTPRDAVARLESMIMMGPVNMPILTIRRNIASAINLIVQVSRMNDGSRKLSHISEIMGMEGDNVILQDIFSF 365 A_act TLEDTAELRLEQPHVVRLETRLAGVEHTGEVTMQDLVINALRMRPERIIVGECRGGEAFQMLQAMNTGHDGSMSTLHANSPRDATSRLESMVMMSNASLPLEAIRRNISSAVNIIVQASRLNDGSRKIMNITEVMGMENGQIVLQDMFSY 377 H_aph TLEDTAELRLEQPHVVRLETRLAGVEHTGEVTMKDLVINALRMRPERIIVGECRGGEAFQMLQAMNTGHDGSMSTLHANSPRDATSRLESMVMMSNATLPLEAIRRNIASAVNIIVQASRLNDGSRKIVNITEIMGMENGQIVLQDIFSY 378 P_mul TLEDTAELRLEQPHVVRLETRLAGVERTGEITMQDLVINALRMRPERIIVGECRGGEAFQMLQAMNTGHDGSMSTLHANSPRDATARLESMVMMSNASLPLEAIRRNIASAVNIIVQASRLNDGSRKIMNITELMGMENGQIVMQDIFSY 378 H_duc TLEDTAELRLEQPHVVRLETRLAGVERTGEITMQDLVINALRMRPERIIVGECRGAEAFQMLQAMNTGHDGSMSTLHANTPRDATARLESMVMMSNASLPLEAIRRNIASAVNIIIQASRLNDGSRKVMNITEVMGMENGQIVLQDIFSF 378 A_pleur TLEDTAELRLEQPHVVRLETRLAGVERTGEISMQDLVINALRMRPERIIVGECRGAEAFQMLQAMNTGHDGSMSTLHANSPRDALARLESMVMMSNASLPLEAIRRNIASAVNIIIQASRLNDGSRKVTNITEVMGMENGQIVLQDIFSY 378 V_vulnI8_11 TIEDAAELSLTQPHWIQLETRTASSEGTGAVTVRDLVKNALRMRPDRIILGEVRGAEAFDMLQAMNTGHDGSLCTLHANSPADAMLRLENMLMMGAEQIPSAVLRQQISSALDLVVQLERSHDGKRRVTAISAVGGIEQGQIVVHPLFEC 389 V_vulnI6_11 TIEDSAELQLQQPHVVRLETRPPNLEGKGEITQRDLVKNALRMRPDRIVLGEVRGAEAVDMLAAMNTGHDGSLATIHANTPRDALSRVENMFAMAGWNISTKNLRAQIASAIHLVVQMERQEDGKRRMVSIQEINGMEGEIITMSEIFHF 450 ruler
17 Displaying Sequence Data Displaying Information Take care with your choice of fixed or variable width fonts. Use fonts carefully and consistently. Avoid using fonts arbitrarily. Use black or dark text against a white or very light background (no more than 20% color) to maximize comprehension. Avoid text that blends with a background, and be cautious in using light text on a dark background. Use shading, case, bold, italic or color when appropriate, to add emphasis, contrast, or draw attention to a feature. Avoid displays in which everything blends together or lacks contrast. Align items to each other to establish a visual connection. Related items should be grouped in close proximity. Avoid simply placing items arbitrarily. Use color logically and aesthetically. Avoid the overuse of color. References The Mac is Not a Typewriter by Robin Williams The Non-Designer s Design Book by Robin Williams The Visual Display of Quantitiative Information by Edward R. Tufte Type & Layout by Colin Wheildon
G4120: Introduction to Computational Biology
 ICB Fall 2004 G4120: Introduction to Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2004 Oliver Jovanovic, All Rights Reserved. Alignment Alignment
ICB Fall 2004 G4120: Introduction to Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2004 Oliver Jovanovic, All Rights Reserved. Alignment Alignment
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Lecture 5: Sequence Analysis March 6, 2017
 ICQB Introduction to Computational & Quantitative Biology (G4120) Spring 2017 Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology & Immunology Sequence Analysis Sequence analysis involves
ICQB Introduction to Computational & Quantitative Biology (G4120) Spring 2017 Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology & Immunology Sequence Analysis Sequence analysis involves
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT
 3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
Sara C. Madeira. Universidade da Beira Interior. (Thanks to Ana Teresa Freitas, IST for useful resources on this subject)
") Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
CONCEPT OF SEQUENCE COMPARISON. Natapol Pornputtapong 18 January 2018
 CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
Bioinformatics. Dept. of Computational Biology & Bioinformatics
 Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Single alignment: Substitution Matrix. 16 march 2017
 Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I)
 Sequence pairwise alignment (I)") CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
Sequence Database Search Techniques I: Blast and PatternHunter tools
 Sequence Database Search Techniques I: Blast and PatternHunter tools Zhang Louxin National University of Singapore Outline. Database search 2. BLAST (and filtration technique) 3. PatternHunter (empowered
Sequence Database Search Techniques I: Blast and PatternHunter tools Zhang Louxin National University of Singapore Outline. Database search 2. BLAST (and filtration technique) 3. PatternHunter (empowered
Biochemistry 324 Bioinformatics. Pairwise sequence alignment
 Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Basic Local Alignment Search Tool
 Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Pairwise & Multiple sequence alignments
 Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Using Bioinformatics to Study Evolutionary Relationships Instructions
 3 Using Bioinformatics to Study Evolutionary Relationships Instructions Student Researcher Background: Making and Using Multiple Sequence Alignments One of the primary tasks of genetic researchers is comparing
3 Using Bioinformatics to Study Evolutionary Relationships Instructions Student Researcher Background: Making and Using Multiple Sequence Alignments One of the primary tasks of genetic researchers is comparing
InDel 3-5. InDel 8-9. InDel 3-5. InDel 8-9. InDel InDel 8-9
 Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Bioinformatics Exercises
 Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment
 Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Multiple sequence alignment
 Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
Practical considerations of working with sequencing data
 Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment
 Pairwise sequence alignment") Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Sequence Alignment (chapter 6)
") Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
BLAST Database Searching. BME 110: CompBio Tools Todd Lowe April 8, 2010
 BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
Homology and Information Gathering and Domain Annotation for Proteins
 Homology and Information Gathering and Domain Annotation for Proteins Outline Homology Information Gathering for Proteins Domain Annotation for Proteins Examples and exercises The concept of homology The
Homology and Information Gathering and Domain Annotation for Proteins Outline Homology Information Gathering for Proteins Domain Annotation for Proteins Examples and exercises The concept of homology The
Alignment & BLAST. By: Hadi Mozafari KUMS
 Alignment & BLAST By: Hadi Mozafari KUMS SIMILARITY - ALIGNMENT Comparison of primary DNA or protein sequences to other primary or secondary sequences Expecting that the function of the similar sequence
Alignment & BLAST By: Hadi Mozafari KUMS SIMILARITY - ALIGNMENT Comparison of primary DNA or protein sequences to other primary or secondary sequences Expecting that the function of the similar sequence
08/21/2017 BLAST. Multiple Sequence Alignments: Clustal Omega
 BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
BMI/CS 776 Lecture #20 Alignment of whole genomes. Colin Dewey (with slides adapted from those by Mark Craven)
") BMI/CS 776 Lecture #20 Alignment of whole genomes Colin Dewey (with slides adapted from those by Mark Craven) 2007.03.29 1 Multiple whole genome alignment Input set of whole genome sequences genomes diverged
BMI/CS 776 Lecture #20 Alignment of whole genomes Colin Dewey (with slides adapted from those by Mark Craven) 2007.03.29 1 Multiple whole genome alignment Input set of whole genome sequences genomes diverged
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT
 5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
Bioinformatics and BLAST
 Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Bioinformatics Definitions The use of computational
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Bioinformatics Definitions The use of computational
Introduction to Bioinformatics
 Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Introduction to protein alignments
 Introduction to protein alignments Comparative Analysis of Proteins Experimental evidence from one or more proteins can be used to infer function of related protein(s). Gene A Gene X Protein A compare
Introduction to protein alignments Comparative Analysis of Proteins Experimental evidence from one or more proteins can be used to infer function of related protein(s). Gene A Gene X Protein A compare
MegAlign Pro Pairwise Alignment Tutorials
 MegAlign Pro Pairwise Alignment Tutorials All demo data for the following tutorials can be found in the MegAlignProAlignments.zip archive here. Tutorial 1: Multiple versus pairwise alignments 1. Extract
MegAlign Pro Pairwise Alignment Tutorials All demo data for the following tutorials can be found in the MegAlignProAlignments.zip archive here. Tutorial 1: Multiple versus pairwise alignments 1. Extract
Similarity or Identity? When are molecules similar?
 Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Alignment principles and homology searching using (PSI-)BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)
BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)") Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Computational Biology
 Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Homology. and. Information Gathering and Domain Annotation for Proteins
 Homology and Information Gathering and Domain Annotation for Proteins Outline WHAT IS HOMOLOGY? HOW TO GATHER KNOWN PROTEIN INFORMATION? HOW TO ANNOTATE PROTEIN DOMAINS? EXAMPLES AND EXERCISES Homology
Homology and Information Gathering and Domain Annotation for Proteins Outline WHAT IS HOMOLOGY? HOW TO GATHER KNOWN PROTEIN INFORMATION? HOW TO ANNOTATE PROTEIN DOMAINS? EXAMPLES AND EXERCISES Homology
Introduction to Bioinformatics Online Course: IBT
 Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
Comparing whole genomes
 BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
Sequence analysis and Genomics
 Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
DATA ACQUISITION FROM BIO-DATABASES AND BLAST. Natapol Pornputtapong 18 January 2018
 DATA ACQUISITION FROM BIO-DATABASES AND BLAST Natapol Pornputtapong 18 January 2018 DATABASE Collections of data To share multi-user interface To prevent data loss To make sure to get the right things
DATA ACQUISITION FROM BIO-DATABASES AND BLAST Natapol Pornputtapong 18 January 2018 DATABASE Collections of data To share multi-user interface To prevent data loss To make sure to get the right things
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
In-Depth Assessment of Local Sequence Alignment
 2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
Protein function prediction based on sequence analysis
 Performing sequence searches Post-Blast analysis, Using profiles and pattern-matching Protein function prediction based on sequence analysis Slides from a lecture on MOL204 - Applied Bioinformatics 18-Oct-2005
Performing sequence searches Post-Blast analysis, Using profiles and pattern-matching Protein function prediction based on sequence analysis Slides from a lecture on MOL204 - Applied Bioinformatics 18-Oct-2005
Ch. 9 Multiple Sequence Alignment (MSA)
") Ch. 9 Multiple Sequence Alignment (MSA) - gather seqs. to make MSA - doing MSA with ClustalW - doing MSA with Tcoffee - comparing seqs. that cannot align Introduction - from pairwise alignment to MSA -
Ch. 9 Multiple Sequence Alignment (MSA) - gather seqs. to make MSA - doing MSA with ClustalW - doing MSA with Tcoffee - comparing seqs. that cannot align Introduction - from pairwise alignment to MSA -
Copyright 2000 N. AYDIN. All rights reserved. 1
 Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Sequence Alignments. Dynamic programming approaches, scoring, and significance. Lucy Skrabanek ICB, WMC January 31, 2013
 Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Collected Works of Charles Dickens
 Collected Works of Charles Dickens A Random Dickens Quote If there were no bad people, there would be no good lawyers. Original Sentence It was a dark and stormy night; the night was dark except at sunny
Collected Works of Charles Dickens A Random Dickens Quote If there were no bad people, there would be no good lawyers. Original Sentence It was a dark and stormy night; the night was dark except at sunny
BLAST. Varieties of BLAST
 BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
Effects of Gap Open and Gap Extension Penalties
 Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Heuristic Alignment and Searching
 3/28/2012 Types of alignments Global Alignment Each letter of each sequence is aligned to a letter or a gap (e.g., Needleman-Wunsch). Local Alignment An optimal pair of subsequences is taken from the two
3/28/2012 Types of alignments Global Alignment Each letter of each sequence is aligned to a letter or a gap (e.g., Needleman-Wunsch). Local Alignment An optimal pair of subsequences is taken from the two
Bioinformatics tools for phylogeny and visualization. Yanbin Yin
 Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
CS612 - Algorithms in Bioinformatics
 Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Biology Tutorial. Aarti Balasubramani Anusha Bharadwaj Massa Shoura Stefan Giovan
 Biology Tutorial Aarti Balasubramani Anusha Bharadwaj Massa Shoura Stefan Giovan Viruses A T4 bacteriophage injecting DNA into a cell. Influenza A virus Electron micrograph of HIV. Cone-shaped cores are
Biology Tutorial Aarti Balasubramani Anusha Bharadwaj Massa Shoura Stefan Giovan Viruses A T4 bacteriophage injecting DNA into a cell. Influenza A virus Electron micrograph of HIV. Cone-shaped cores are
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Background: comparative genomics. Sequence similarity. Homologs. Similarity vs homology (2) Similarity vs homology. Sequence Alignment (chapter 6)
 Similarity vs homology. Sequence Alignment (chapter 6)") Sequence lignment (chapter ) he biological problem lobal alignment Local alignment Multiple alignment Background: comparative genomics Basic question in biology: what properties are shared among organisms?
Sequence lignment (chapter ) he biological problem lobal alignment Local alignment Multiple alignment Background: comparative genomics Basic question in biology: what properties are shared among organisms?
Phylogenies Scores for Exhaustive Maximum Likelihood and Parsimony Scores Searches
 Int. J. Bioinformatics Research and Applications, Vol. x, No. x, xxxx Phylogenies Scores for Exhaustive Maximum Likelihood and s Searches Hyrum D. Carroll, Perry G. Ridge, Mark J. Clement, Quinn O. Snell
Int. J. Bioinformatics Research and Applications, Vol. x, No. x, xxxx Phylogenies Scores for Exhaustive Maximum Likelihood and s Searches Hyrum D. Carroll, Perry G. Ridge, Mark J. Clement, Quinn O. Snell
Journal of Proteomics & Bioinformatics - Open Access
 Abstract Methodology for Phylogenetic Tree Construction Kudipudi Srinivas 2, Allam Appa Rao 1, GR Sridhar 3, Srinubabu Gedela 1* 1 International Center for Bioinformatics & Center for Biotechnology, Andhra
Abstract Methodology for Phylogenetic Tree Construction Kudipudi Srinivas 2, Allam Appa Rao 1, GR Sridhar 3, Srinubabu Gedela 1* 1 International Center for Bioinformatics & Center for Biotechnology, Andhra
Ensembl focuses on metazoan (animal) genomes. The genomes currently available at the Ensembl site are:
 genomes. The genomes currently available at the Ensembl site are:") Comparative genomics and proteomics Species available Ensembl focuses on metazoan (animal) genomes. The genomes currently available at the Ensembl site are: Vertebrates: human, chimpanzee, mouse, rat,
Comparative genomics and proteomics Species available Ensembl focuses on metazoan (animal) genomes. The genomes currently available at the Ensembl site are: Vertebrates: human, chimpanzee, mouse, rat,
Sequences, Structures, and Gene Regulatory Networks
 Sequences, Structures, and Gene Regulatory Networks Learning Outcomes After this class, you will Understand gene expression and protein structure in more detail Appreciate why biologists like to align
Sequences, Structures, and Gene Regulatory Networks Learning Outcomes After this class, you will Understand gene expression and protein structure in more detail Appreciate why biologists like to align
Phylogenetic analyses. Kirsi Kostamo
 Phylogenetic analyses Kirsi Kostamo The aim: To construct a visual representation (a tree) to describe the assumed evolution occurring between and among different groups (individuals, populations, species,
Phylogenetic analyses Kirsi Kostamo The aim: To construct a visual representation (a tree) to describe the assumed evolution occurring between and among different groups (individuals, populations, species,
Motivating the need for optimal sequence alignments...
 1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
Pairwise sequence alignment
 Department of Evolutionary Biology Example Alignment between very similar human alpha- and beta globins: GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKL G+ +VK+HGKKV A+++++AH+D++ +++++LS+LH KL GNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKL
Department of Evolutionary Biology Example Alignment between very similar human alpha- and beta globins: GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKL G+ +VK+HGKKV A+++++AH+D++ +++++LS+LH KL GNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKL
Fundamentals of database searching
 Fundamentals of database searching Aligning novel sequences with previously characterized genes or proteins provides important insights into their common attributes and evolutionary origins. The principles
Fundamentals of database searching Aligning novel sequences with previously characterized genes or proteins provides important insights into their common attributes and evolutionary origins. The principles
Sequence Analysis and Databases 2: Sequences and Multiple Alignments
 1 Sequence Analysis and Databases 2: Sequences and Multiple Alignments Jose María González-Izarzugaza Martínez CNIO Spanish National Cancer Research Centre (jmgonzalez@cnio.es) 2 Sequence Comparisons:
1 Sequence Analysis and Databases 2: Sequences and Multiple Alignments Jose María González-Izarzugaza Martínez CNIO Spanish National Cancer Research Centre (jmgonzalez@cnio.es) 2 Sequence Comparisons:
Whole Genome Alignments and Synteny Maps
 Whole Genome Alignments and Synteny Maps IINTRODUCTION It was not until closely related organism genomes have been sequenced that people start to think about aligning genomes and chromosomes instead of
Whole Genome Alignments and Synteny Maps IINTRODUCTION It was not until closely related organism genomes have been sequenced that people start to think about aligning genomes and chromosomes instead of
USING BLAST TO IDENTIFY PROTEINS THAT ARE EVOLUTIONARILY RELATED ACROSS SPECIES
 USING BLAST TO IDENTIFY PROTEINS THAT ARE EVOLUTIONARILY RELATED ACROSS SPECIES HOW CAN BIOINFORMATICS BE USED AS A TOOL TO DETERMINE EVOLUTIONARY RELATIONSHPS AND TO BETTER UNDERSTAND PROTEIN HERITAGE?
USING BLAST TO IDENTIFY PROTEINS THAT ARE EVOLUTIONARILY RELATED ACROSS SPECIES HOW CAN BIOINFORMATICS BE USED AS A TOOL TO DETERMINE EVOLUTIONARY RELATIONSHPS AND TO BETTER UNDERSTAND PROTEIN HERITAGE?
DNA and protein databases. EMBL/GenBank/DDBJ database of nucleic acids
 Database searches 1 DNA and protein databases EMBL/GenBank/DDBJ database of nucleic acids 2 DNA and protein databases EMBL/GenBank/DDBJ database of nucleic acids (cntd) 3 DNA and protein databases SWISS-PROT
Database searches 1 DNA and protein databases EMBL/GenBank/DDBJ database of nucleic acids 2 DNA and protein databases EMBL/GenBank/DDBJ database of nucleic acids (cntd) 3 DNA and protein databases SWISS-PROT
A greedy, graph-based algorithm for the alignment of multiple homologous gene lists
 A greedy, graph-based algorithm for the alignment of multiple homologous gene lists Jan Fostier, Sebastian Proost, Bart Dhoedt, Yvan Saeys, Piet Demeester, Yves Van de Peer, and Klaas Vandepoele Bioinformatics
A greedy, graph-based algorithm for the alignment of multiple homologous gene lists Jan Fostier, Sebastian Proost, Bart Dhoedt, Yvan Saeys, Piet Demeester, Yves Van de Peer, and Klaas Vandepoele Bioinformatics
The Contribution of Bioinformatics to Evolutionary Thought
 The Contribution of Bioinformatics to Evolutionary Thought A demonstration of the abilities of Entrez, BLAST, and UCSC s Genome Browser to provide information about common ancestry. American Scientific
The Contribution of Bioinformatics to Evolutionary Thought A demonstration of the abilities of Entrez, BLAST, and UCSC s Genome Browser to provide information about common ancestry. American Scientific
Example of Function Prediction
 Find similar genes Example of Function Prediction Suggesting functions of newly identified genes It was known that mutations of NF1 are associated with inherited disease neurofibromatosis 1; but little
Find similar genes Example of Function Prediction Suggesting functions of newly identified genes It was known that mutations of NF1 are associated with inherited disease neurofibromatosis 1; but little
Phylogenetic inference
 Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Tutorial 4 Substitution matrices and PSI-BLAST
 Tutorial 4 Substitution matrices and PSI-BLAST 1 Agenda Substitution Matrices PAM - Point Accepted Mutations BLOSUM - Blocks Substitution Matrix PSI-BLAST Cool story of the day: Why should we care about
Tutorial 4 Substitution matrices and PSI-BLAST 1 Agenda Substitution Matrices PAM - Point Accepted Mutations BLOSUM - Blocks Substitution Matrix PSI-BLAST Cool story of the day: Why should we care about
Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction. Lesser Tenrec (Echinops telfairi)
") Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction Lesser Tenrec (Echinops telfairi) Goals: 1. Use phylogenetic experimental design theory to select optimal taxa to
Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction Lesser Tenrec (Echinops telfairi) Goals: 1. Use phylogenetic experimental design theory to select optimal taxa to
Hands-On Nine The PAX6 Gene and Protein
 Hands-On Nine The PAX6 Gene and Protein Main Purpose of Hands-On Activity: Using bioinformatics tools to examine the sequences, homology, and disease relevance of the Pax6: a master gene of eye formation.
Hands-On Nine The PAX6 Gene and Protein Main Purpose of Hands-On Activity: Using bioinformatics tools to examine the sequences, homology, and disease relevance of the Pax6: a master gene of eye formation.
A Browser for Pig Genome Data
 A Browser for Pig Genome Data Thomas Mailund January 2, 2004 This report briefly describe the blast and alignment data available at http://www.daimi.au.dk/ mailund/pig-genome/ hits.html. The report describes
A Browser for Pig Genome Data Thomas Mailund January 2, 2004 This report briefly describe the blast and alignment data available at http://www.daimi.au.dk/ mailund/pig-genome/ hits.html. The report describes
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program)
") Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Moreover, the circular logic
 Moreover, the circular logic How do we know what is the right distance without a good alignment? And how do we construct a good alignment without knowing what substitutions were made previously? ATGCGT--GCAAGT
Moreover, the circular logic How do we know what is the right distance without a good alignment? And how do we construct a good alignment without knowing what substitutions were made previously? ATGCGT--GCAAGT
Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM)
") Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Overview Multiple Sequence Alignment
 Overview Multiple Sequence Alignment Inge Jonassen Bioinformatics group Dept. of Informatics, UoB Inge.Jonassen@ii.uib.no Definition/examples Use of alignments The alignment problem scoring alignments
Overview Multiple Sequence Alignment Inge Jonassen Bioinformatics group Dept. of Informatics, UoB Inge.Jonassen@ii.uib.no Definition/examples Use of alignments The alignment problem scoring alignments
Sequence Bioinformatics. Multiple Sequence Alignment Waqas Nasir
 Sequence Bioinformatics Multiple Sequence Alignment Waqas Nasir 2010-11-12 Multiple Sequence Alignment One amino acid plays coy; a pair of homologous sequences whisper; many aligned sequences shout out
Sequence Bioinformatics Multiple Sequence Alignment Waqas Nasir 2010-11-12 Multiple Sequence Alignment One amino acid plays coy; a pair of homologous sequences whisper; many aligned sequences shout out
Exercise 5. Sequence Profiles & BLAST
 Exercise 5 Sequence Profiles & BLAST 1 Substitution Matrix (BLOSUM62) Likelihood to substitute one amino acid with another Figure taken from https://en.wikipedia.org/wiki/blosum 2 Substitution Matrix (BLOSUM62)
Exercise 5 Sequence Profiles & BLAST 1 Substitution Matrix (BLOSUM62) Likelihood to substitute one amino acid with another Figure taken from https://en.wikipedia.org/wiki/blosum 2 Substitution Matrix (BLOSUM62)
Investigation 3: Comparing DNA Sequences to Understand Evolutionary Relationships with BLAST
 Investigation 3: Comparing DNA Sequences to Understand Evolutionary Relationships with BLAST Introduction Bioinformatics is a powerful tool which can be used to determine evolutionary relationships and
Investigation 3: Comparing DNA Sequences to Understand Evolutionary Relationships with BLAST Introduction Bioinformatics is a powerful tool which can be used to determine evolutionary relationships and
EBI web resources II: Ensembl and InterPro
 EBI web resources II: Ensembl and InterPro Yanbin Yin http://www.ebi.ac.uk/training/online/course/ 1 Homework 3 Go to http://www.ebi.ac.uk/interpro/training.htmland finish the second online training course
EBI web resources II: Ensembl and InterPro Yanbin Yin http://www.ebi.ac.uk/training/online/course/ 1 Homework 3 Go to http://www.ebi.ac.uk/interpro/training.htmland finish the second online training course
Introduction to Bioinformatics Introduction to Bioinformatics
 Dr. rer. nat. Gong Jing Cancer Research Center Medicine School of Shandong University 2012.11.09 1 Chapter 4 Phylogenetic Tree 2 Phylogeny Evidence from morphological ( 形态学的 ), biochemical, and gene sequence
Dr. rer. nat. Gong Jing Cancer Research Center Medicine School of Shandong University 2012.11.09 1 Chapter 4 Phylogenetic Tree 2 Phylogeny Evidence from morphological ( 形态学的 ), biochemical, and gene sequence
Comparative Bioinformatics Midterm II Fall 2004
 Comparative Bioinformatics Midterm II Fall 2004 Objective Answer, part I: For each of the following, select the single best answer or completion of the phrase. (3 points each) 1. Deinococcus radiodurans
Comparative Bioinformatics Midterm II Fall 2004 Objective Answer, part I: For each of the following, select the single best answer or completion of the phrase. (3 points each) 1. Deinococcus radiodurans
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences
 Analysis of Biological Sequences") First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
Sequence Analysis '17- lecture 8. Multiple sequence alignment
 Sequence Analysis '17- lecture 8 Multiple sequence alignment Ex5 explanation How many random database search scores have e-values 10? (Answer: 10!) Why? e-value of x = m*p(s x), where m is the database
Sequence Analysis '17- lecture 8 Multiple sequence alignment Ex5 explanation How many random database search scores have e-values 10? (Answer: 10!) Why? e-value of x = m*p(s x), where m is the database
Sequence Analysis 17: lecture 5. Substitution matrices Multiple sequence alignment
 Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of