Molecular Modeling and In Silico Evaluation of Novel Pyridazinones Derivatives as Anticonvulsant Agents
|
|
|
- Coleen Copeland
- 6 years ago
- Views:
Transcription
1 imedpub Journals Journal of In Silico & In Vitro Pharmacology Abstract Molecular Modeling and In Silico Evaluation of Novel Pyridazinones Derivatives as Anticonvulsant Agents Context: Virtual screening techniques or computational methods are used for the drug discovery and development. bjective: In search for the safer and effective anticonvulsant agents, docking study on pyridazinone derivatives was performed to potentiate GABA mediated chlorine channel opening. Methods: A total of eighteen compounds were screened for their anticonvulsant activity using molecular docking inside the ligand binding domain of PDB ID 2Q1Q using Molegro Virtual docker. QikProp 3.4 & molinspiration software were also used to evaluate drug likeliness and to perform comparative bioactivity analysis for all the pyridazinone derivatives. Results: ut of eighteen compounds, (I) showed highest Mol dock score and (XVIII) formed maximum number of hydrogen bond interactions. xygen atom of the Nitro group on the biphenyl ring of compound (XV) formed a strong hydrogen bond with with a distance 2.33A. Mol dock score of diazepam (standard drug) was Conclusions: Based on the results of mol dock score and number of hydrogen bond interactions, compounds (XVIII) and (XIII) observed to be the most potent compounds. Keywords: Convulsant; ADME; Docking; Heterocyclic; Pyridazinone. Husain A 1, Khokra SL 2, Thakur P 2, Choudhary D 2, Kohli S 1, Ahmad A 3 and Khan SA 4 1 Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Jamia Hamdard (Hamdard University), New Delhi , India. 2 Institute of Pharmaceutical Sciences, Kurukshetra University, Haryana , India. 3 Health Information Technology Department, Jeddah Community College, King Abdulaziz University, Jeddah, Saudi Arabia. 4 Department of Pharmacy, man Medical College, Muscat, Sultanate of man. Corresponding author: Asif Husain asifhusain@yahoo.com Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Jamia Hamdard (Hamdard University), New Delhi , Introduction The term In silico or computational methods are virtual screening techniques used for the discovery of novel molecules of potential pharmaceutical interests and for advancement in therapeutics. The in silico studies considered as complementary to in vivo and in vitro biological studies are performed by using a computer and are playing increasingly larger and more important role in drug discovery and development. Computational methods include, docking, homology modeling, quantitative structure activity relationships (QSARs), similarity and pharmacophore searching, virtual ligand screening, data mining, and data analysis tools [1]. These tools are becoming increasingly popular in drug design and the last decade has witnessed an upsurge in the development and application of computational methods applied to the drug development [2]. These methods are used in the drug discovery and optimization of novel molecules having potential affinity and specificity for the selected therapeutic targets. The pharmacokinetic (PK) properties of a drug molecule such as Absorption, Distribution, Metabolism, Excretion & toxicity (ADME-T) depend more or less on the chemical descriptors of molecule and have become quite important in the drug-discovery process. Prediction and estimation of PK properties by using computational methods have become increasingly important in drug selection, promotion process and also are promising tools for the fast screening of potential drug candidate [3]. Epilepsy is a chronic neurological disorder that is characterized by recurrent unprovoked seizures [4]. Traditionally, pharmacological strategies 1
2 Journal of In Silico & ARCHIVS In Vitro Pharmacology for the treatment of epilepsy are aimed at suppressing the initiation or propagation of seizures rather than the treatment of the underling processes that lead to epilepsy [5]. Because of this approach, the epilepsy is usually controlled but not cured with the anticonvulsants drugs. The correct approach in the treatment of epilepsy should be to identify new targets through better understanding of molecular mechanism of epilepsy in first place. Second strategy should aim at modification of already existing drugs and formulations [6]. Anticonvulsant agents act by more than one mechanism of action which include prolong inactivation of the ion channel or potentiating neurotransmitter system [7-9]. In fact some of the clinically used anticonvulsant drugs have not been linked with the specific site in the brain, and their exact mechanism of action remain unknown [10]. In the past, conventional screening and/or structural modification methods were used for the discovery of novel anticonvulsant agents rather than a mechanism-driven drug design process. Therefore, drug identification was generally carried out on the basis of seizures type i.e. in vivo screening tests rather than etiology of the disease. In recent years, considerable emphasis has been placed on the relation between epileptic seizures and neurotransmitter in the brain. Augmenting the inhibition of neurotransmission or diminishing the excitement of transmission provides a great approach to the design of anticonvulsant and anti-epileptic drugs. γ-aminobutyric acid (GABA) is a good example of inhibitory neurotransmission. GABA being water soluble is difficult to pass through the blood-brain barrier (BBB) and thus penetrates poorly into the brain; therefore, it cannot be used as an anti-epileptic in clinics. Hence, to improve its lipid solubility or ability to cross the BBB, a series of arylpyridazine derivatives of GABA were designed [11,12]. Pyridazinones are the derivatives of pyridazine possessing a magic azomethine (-NH-N=CH-) moiety. The pyridazinone are six membered heterocyclic compounds that also contains a cyclic amide moiety in their ring structure that plays an important role in exhibiting various pharmacological activity ranging from cardiovascular properties, anti-inflammatory, antidiabetic, analgesic, anti-aids, anti-cancer, anti-microbial and anticonvulsant activities [13,14]. Thus pyridazinone appear to be potential drug candidates for the anticonvulsant therapy. In order to develop potent and safe anticonvulsant drugs, many researchers have synthesized the different pyridazine and pyridazinone derivatives and evaluated for their anticonvulsant activity in different animal models of seizures [15]. They observed that presence of a phenyl substitution on pyridazinone ring with electron withdrawing group possess good activity against tonicclonic seizures. Using this background information and in line of our current interest in the protein ligand binding, we describe herein the in silico study of various hypothetical pyridazinones and their interaction with facilitator of GABA transmission by computational docking method. The compounds found to have high Mole Dock Score and formed maximum number of H-bond interaction were synthesized and evaluated for their anticonvulsant activities. However computational data based on docking studies is presented in this research paper. Materials and methods Docking is the computational method, which predicts the 2 preferred orientation of one molecule to a second, when bound to each other to form a stable complex. Docking study was performed with a set of hypothetical pyridazinone derivatives using Molegro Virtual docker MVD on X-ray structures of GABA (PDB ID: 2Q1Q) downloaded from the protein data bank (PDB). Ligand preparation The molecules were draen in MarvinSketch After converting the 2D molecules into 3D, the conformational energy of molecules was minimized. The resulting structures were saved in MarvinSketch as MDL Molfile (*.mol). The simplest use of MarvinSketch produces a single, low energy with 3D structure. Finally, the prepared 3D structures of molecules were imported by dragging or dropping a molecule structure file in the workspace. Protein preparation and detecting cavities of protein molecules The GABA facilitator (PDB ID 2QIQ) X-ray structures were accessed from the protein data bank (PDB) and imported. The protein structures were prepared using the protein preparation wizard in MVD. In this step, bond order were assigned, all hydrogen s in the structure were added, and bonds to metals were deleted and adjust the formal charge on the metal and the neighbouring atoms and deleted waters that were more than 5A specific distance. The energy of imported molecules was minimized using Ligand Energy inspector. The energy minimization supports in stability of molecules to be imported. Next, the protein surface was created using protein preparation. This step helps to inspect and change the protonation state for the residues. In order to determine the potential binding sites, a cavity prediction algorithm was performed. Executing a docking set up through docking wizard panel Molegro searches for suitable interaction between one or more ligand molecules and the receptor. Further, all ligands were selected and docking was performed through the docking wizard panel. After this step docking results were imported. Various orientation (or poses) of docked compounds were analysed by determination of Mol Dock Score and H-bond interaction. Compounds were arranged according to their Mol Dock scores and were visualized inside the pocket to view their fitting and closure to main residues. Molecular docking studies revealed further insight into the nature of interactions between the compounds and the active site amino acids to rationalize the obtained biological results. By this docking study we came to know that most of our designed ligands are interacting to GABA facilitated target with sufficient selectively and specificity. The docking analysis is done and the results are presented in the form of image given in (Figure 1-3), furthermore MolDock score and hydrogen bond interactions between receptor and ligands are presented in the Table 1 (see supplementary material). Ligand based ADME&Toxicity studies ADME-T properties were calculated using Qikprop 3.6 tool of Schrodinger 2014 & molinspiration. All the structures

![Tables 3-5, additionally showed drug-like characteristics based on Lipinski s rule of 5 [16].](/docs-images/77/75688577/images/3-1.jpg "Qikprop predicts physically significant descriptors and pharmaceutically relevant properties like absorption, distribution, metabolism, excretion and toxicity (ADMET) & acceptability of compounds")


3 Journal of In Silico & ARCHIVS In Vitro Pharmacology Figure 1 Binding affinity of synthesized compound in specific cavity. Figure 2 Superimposition of synthesized compounds with internal ligand in active site. showed significant values for the properties analyzed. Tables 3-5, additionally showed drug-like characteristics based on Lipinski s rule of 5 [16]. Qikprop predicts physically significant descriptors and pharmaceutically relevant properties like absorption, distribution, metabolism, excretion and toxicity (ADMET) & acceptability of compounds based on Lipinski s rule of five, which is essential to ensure drug-like pharmacokinetic profile while using rational drug design & Molinspiration, an online tool, used to perform QSAR studies in order to identify potential activators of biological targets. Results and Discussion Docking studies revealed that the all the compounds had good potentiating power towards GABA mediated Chlorine channel opening. Docking studies on hypothetically designed compounds of pyridazinone derivatives showed good docking results. Molegro Virtual Docker allows the flexible docking of ligand into its site of action. In the analysis of docking results, we found that the hypothetically design compounds selected for docking studies were able to interact with the hydrophobic pocket of the protein efficiently and shows good anticonvulsant activity. Substitution on phenyl ring with electron withdrawing group also showed good results. Most important feature of docking is the logical interaction of the ligand with the putative-binding site of enzyme. The molecules were built in MarvinSketch The simplest use of MarvinSketch produces a single, low energy with 3D structure. The energy minimization supports in stability of molecules to be imported. Each search was continued until the global energy minima were carried. The protein structures were prepared using the protein preparation wizard in MVD and optimization of hydrogen bond network was carried out. The favourable interaction between one or more ligand molecules and a receptor molecule was carried out by detecting cavity where ever required, to ensure that possible activities were not missed. The process of evaluating a particular pose was achieved by counting the number of suitable interactions that might be hydrogen, hydrophobic or electrostatic bonding. 3


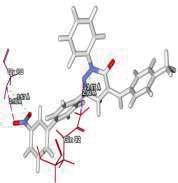
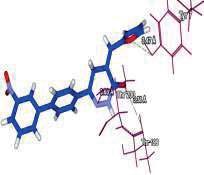
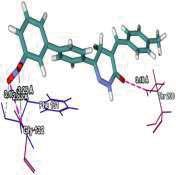
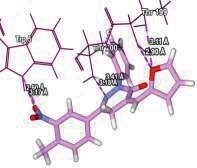
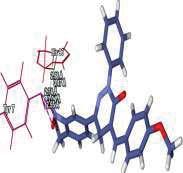
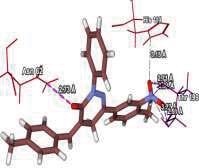
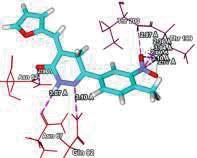
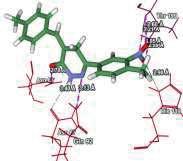
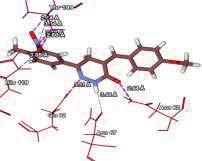
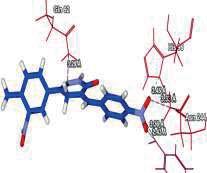
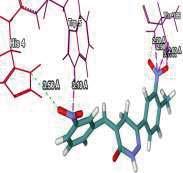
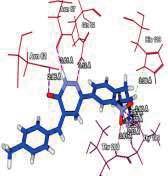
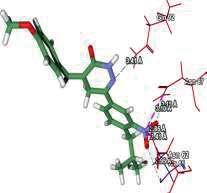
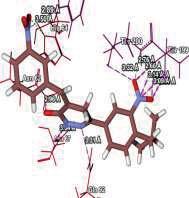
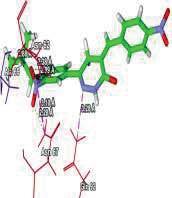
4 Journal of In Silico & ARCHIVS In Vitro Pharmacology Figure 3 Docking images of pyridazinone derivatives (I-XVIII) showing hydrogen bonds with 2QIQ protein residues. 4
5 Journal of In Silico & ARCHIVS In Vitro Pharmacology Table 1 Structures of all the hypothetical pyridazinone derivatives & standard drug. 2 N R' N N R Compound no. R R (I) C 6 (II) C 6 CH 3 (III) C 6 CH 3 (IV) H (V) H CH 3 (VI) H CH 3 H 3 C 2 N R' N N R (VII) C 6 (VIII) C 6 CH 3 (IX) C 6 CH 3 (X) H (XI) H CH 3 (XII) H CH 3 (XIII) H N 2 (XIV) H N 2 H 3 C 2 N CH 3 R' N N R (XV) H CH 3 5
6 Journal of In Silico & ARCHIVS In Vitro Pharmacology (XVI) H CH 3 (XVII) H N 2 (XVIII) H N 2 Diazepam (Standard drug) H N Cl N Table 2 Mol dock score and bonding interaction of pyridazinone derivatives with amino acids resides using PDB ID: 2Q1Q for determination of anticonvulsant activity. Compound Mol Dock Score No. of H-bond Diazepam (Standard drug) (I) (II) (III) (IV) (V) (VI) (VII) H-bond distance Angstrom Amino acid involved Gly132 Phe95 Phe66 Asn244 Asn244 Phe95 Gly132 Gly132 Phe131 Gly132 Gly132 Asn244 His96 Trp5 Trp5 Structural feature N N N N - N of pyridazinone of ketone N of pyridazinone N of pyridazinone N of pyridazinone N of pyridazinone of furfuryl N of pyridazinone of ketone of CH 3 of CH 3 N of pyridazinone of furfuryl of furfuryl N of pyridazinone N of pyridazinone 6
7 Journal of In Silico & ARCHIVS In Vitro Pharmacology (VIII) (IX) (X) (XI) (XII) (XIII) (XIV) (XV) His96 His96 Asp62 His119 His119 The199 Gln62 His119 Asn244 His96 Trp5 His4 His119 of ketone of ketone of ketone N of pyridazinone N of pyridazinone of ketone N of pyridazinone N of pyridazinone of ketone N of pyridazinone N of pyridazinone N of pyridazinone of ketone N of pyridazinone N of pyridazinone 7
8 Journal of In Silico & ARCHIVS In Vitro Pharmacology (XVI) (XVII) (XVIII) His64 Trp5 Ala65 N of pyridazinone of ketone N of pyridazinone Based on this analysis, the residues, Asp62,, Ala65, Phe66,,, Phe95, His119, Phe131, Gly132,,, Asn244, Trp6, Glu7, Thr89, Leu78, Arg67, Arg69, Ser90 and Asp47 were predicted to interact with GABA receptor and to act as functional residues. Mol Dock Score of Standard Compound Diazepam was Diazepam formed four hydrogen bonds interactions with two amino acids which are & and the distance of respective amino acid is 3.00 (A ), 3.48(A ), 3.54 (A ) and 3.21(A ). ut of eighteen compounds, Compound (I) exhibited relatively comparable binding affinity with highest Mol Dock Score of comparable to standard compound. Compound (XVIII) form maximum hydrogen bond interactions i.e. eleven hydrogen bond with six amino acid which are Trp5,,, His64, & the distance of respective amino acid are 2.89(A ), 2.96(A ), 3.04(A ), 3.31(A ), 3.32(A ), 2.76(A ), 2.66(A ), 2.77(A ), 3.14(A ) & 3.09 (A ). xygen of the Nitro group on the biphenyl ring of compound (I) formed a strong hydrogen bond with with a distance 2.33(A ). Some other docked molecule also showed good Mol dock Score and H-bond interactions viz., (VII), (VIII), (X), (XII), (XIII), (XIV), (XVII), and (XVIII). n the basis of docking studies most potent anticonvulsant compounds in our study were found to be (I) and (XVIII), because these showed highest Mol dock Score and form maximum number of hydrogen bond interactions (Table 2). The compounds that had highest Mol Dock Score and minimum energy can be used for further drug designing and also provides a way to synthesize new potent compounds in laboratory. Drug likeliness, stars, log P, log S, molecular weight, QPlogBB, percent human oral absorption, PSA, SASA etc. may be used to estimate the compound s overall potential to achieve a ligand as potential drug candidate. The most important pharmaceutical descriptor in qikprop is stars, which are the combination of all of all properties. A few numbers of stars reflects more drug likeness in the range. ut of eighteen deigned compounds, six compounds showed 0 values, and eight compounds showed 1 value of stars. Range of 8 some other important descriptor were, molecular weight of the designed compounds fall in between the range Daltons, SASA range were , estimated number of HBdonor by the solute to water molecules in the aqueous solutions were , Estimated number of accept HB by the solute to water in aqueous solutions were , predicted QP log P CT were , predicted QP log P W were , predicted QP log P /W were , predicted QP logs were , predicted QP logbb were , predicted human oral absorption were and predicted PSA were ut of eighteen deigned compounds, twelve compounds showed zero Lipinski violation and remaining compounds showed one Lipinski violation satisfying pharmacological properties of 95% available drugs high to medium estimated oral absorption availability. When the Lipinski violation is more than one at that time there is problem in oral bioavailability. The deigned compounds did not show toxic groups. All the analogs were neutralized before being used by Qikprop & molinspiration. All the structures showed significant values for the properties analyzed Table 3 and showed drug-like characteristics based on Lipinski s rule of 5. The ligand based ADME-T values of design compound inhibitors are given in Table 4. The compounds obey Lipinski rule of five for molecular weight (mol MW) less than 500, solubility (QPlogS) greater than-4 number of ratable bond (#rotor) less than 10, and number of hydrogen bond donor and accepter (Hb-doner and accepter) are less than 5 and less than 10 respectively but show violations for partition coefficient between octanol and water (logp (/W)) which is found between 2 to 6.5. Brain/blood partition coefficient (QPlogBB) parameter indicated the ability of the drug to pass through the blood brain barrier which is mandatory for enhancement of GABA-ergic inhibition. Whereas QPPMDCK predicted apparent MDCK cell permeability in nm/s. MDCK cells are considered to be a good mimic for the blood brain barrier. Higher the value of MDCK cell, higher the
9 Journal of In Silico & ARCHIVS In Vitro Pharmacology Table 3 Results of ligand based ADME-T studies of proposed pyridazinone derivatives. Compounds # star #rotor MW QPLog Khsa SASA Volume Donor HB Accpt HB QP log P CT QP log P W (I) (II) (III) (IV) (V) (IV) (VI) (VIII) (IX) (X) (XI) (XII) (XIII) (XIV) (XV) (XVI) (XVII) (XVIII) MW: Molecular weight; QPLog Khsa: Prediction of binding to human serum albumin; SASA: Total solvent accessible surface area; donor HB: number of hydrogen bonds donated by solute to water molecule; Accpt HB: number of hydrogen bonds accepted by solute from water molecule; QP logp CT : Predicted octanol/gas partition coefficient; QP log P W : Predicted water/gas partition coefficient. Table 4 Results of ligand based ADME-T studies of proposed pyridazinone derivatives Compounds QP log QP HUMAN RAL QP LogBB QPPCaco QPPMDCK QPLog Kp P /W Log S ABSRPTIN % PSA RULE F 5 HA (I) (II) (III) (IV) (V) (VI) (VII) (VIII) (IX) (X) (XI) (XII) (XIII) (XIV) (XV) (XVI) (XVII) (XVIII) QP log P /W : Predicted octanol/water partition coefficient; QPLog S: Predicted aqueous solubility, log S; QP LogBB: Predicted brain/blood partition coefficient; QPPCaco: Predicted apparent Caco-2 cell permeability in nm/sec; QPPMDCK: Predicted apparent MDCK cell permeability in nm/sec; QPLog Kp: Predicted skin permeability, log Kp; PSA: Polar surface area cell permeability. ut of eighteen compounds sixteen designed compounds showed acceptable range of QPMDCK, fourteen compounds showed few number of stars viz; (IV), (X), (XI), (XII), (XIII), (XIV) showed zero stars, and (II), (VI), (VII), (VIII), (XV), (XVI), (XVII), (XVIII) showed one stars. Therefore these compounds showed most favorable results for anticonvulsant activity and the compounds that showed acceptable range of all descriptors can be assigned for further drug designing and also synthesize these compounds in laboratory. The most active compounds of our study were (IV), (X), (XI), (XII), (XIII), (XIV), (II), (VI), (VII), (VIII), (XV), (XVI), (XVII), (XVIII) because these designed compounds showed acceptable range of the descriptors. Drug likeness properties by molinspiration also showed good results showed in Table 5. n comparing the relative activity scores of diazepam 9
10 Journal of In Silico & ARCHIVS In Vitro Pharmacology Table 5 Results of drug likeness properties viz; bioactivity score of proposed pyridazinone derivatives by molinspiration. Compound no. GPCR ligand Ion Channel Nuclear receptor Kinase inhibitor Modulator ligand Protease inhibitor Enzyme inhibitor (I) (II) (III) (IV) (V) (VI) (VI) (VIII) (IX) (X) (XI) (XII) (XIII) (XIV) (XV) (XVI) (XVII) (XVIII) Diazepam with (I-XVIII) compounds utilizing the six drug classes, all the compounds showed similar bioactivity score as compared to the standard drug viz; Diazepam, especially in case of GPCR ligand, ions channel modulator, kinases inhibitor and nuclear receptor ligand. Conclusion A brief computational study was carried out over eighteen designed pyridazinone derivatives using various software programs with the goal of identifying potential lead molecules that efficiently bind to the human GABA receptors. Docking studies have helped us to know about the binding modes of hypothetical designed pyridazinone derivatives to determine their GABA facilitator activities. These investigations paved the way for the synthesis of selected compounds, which are more potent and selective GABA facilitator. Moreover, it was also proved from above discussion that geometry of receptor also plays a major role in defining drug action. ur ADME-T studies permit us to evaluate a set of anticonvulsant lead compounds and to assess the parameter that will be essential for further lead optimization efforts. The in silico evaluation confirmed that, the compounds had druglike properties but did not give information of sufficient value to discriminate between compounds. Succeeding lead optimization efforts are warranted to produce a fully optimized anticonvulsant drug candidate ready for preclinical development studies. 10
11 Journal of In Silico & ARCHIVS In Vitro Pharmacology References 1 Ekins S, Mestres J, Testa B (2007) In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br J Pharmacol 152: Ballester GF, Carvajal AF, Gonzalez-Ros JM, Montiel AF (2011) Ionic Channels as Tartgets for Drug Design: A Review on Computational Methods. Pharmaceutics 3: Moroy G, Martiny VY, Vayer P, Villoutreix B, Miteva MA (2012) Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov Today 17: Blume WT, Lüders H, Mizrahi E, Tassinari C, van Emde Boas W, et al. (2001) Glossary of descriptive terminology for ictal semiology: report of the ILAE task force on classification and terminology. Epilepsia 42: Beyenburg S, Bauer J, Reuber M (2004) New drugs for the treatment of epilepsy: a practical approach. Postgrad Med J 80: Edafiogho I, Scott KR (1996) Burgers Medicinal Chemistry and Drug Discovery. John Wiley and Sons, New York, USA, Pp: Cosford ND, Tehrani L, Roppe J, Schweiger E, Smith ND, et al. (2003) 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J Med Chem 46: Jansen M, Dannhardt G (2003) Antagonists and agonists at the glycine site of the NMDA receptor for therapeutic interventions. Eur J Med Chem 38: Czuczwar SJ, Patsalos PN (2001) The new generation of GABA enhancers. Potential in the treatment of epilepsy.cns Drugs 15: Jannuzzi G, Cian P, Fattore C, Gatti G, Bartoli A, et al. (2000) A multicenter randomized controlled trial on the clinical impact of therapeutic drug monitoring in patients with newly diagnosed epilepsy. The Italian TDM Study Group in Epilepsy. Epilepsia 41: Dam M, Bolwig T, Hertz M, Bajorec J, Lomax P, et al. (1984) Does seizure activity produce Purkinje cell loss? Epilepsia 25: Allan RD, Johnston GAR, Kazlauskas R, Tran HW (1983) Synthesis of analogues of gamma-aminobutyric acid. Part 11. Unsaturated and saturated tetronic acid derivatives. J Chem Soc Perkin Katritzky AR, Charles WR (1984) Pergamon press, UK, Pp: Banerjee PS, Sharma PK (2011) New antiepileptic agents: structureactivity relationships. Med Chem Res 21: Rubat C, Coudert P, Albuisson E, Basitde J, Couquelet J, et al. (1992) Synthesis of Mannich bases of arylidenepyridazinones as analgesic agents. J Pharm Sci 81: Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev 46:
ASSESSING THE DRUG ABILITY OF CHALCONES USING IN- SILICO TOOLS
 Page109 IJPBS Volume 5 Issue 1 JAN-MAR 2015 109-114 Research Article Pharmaceutical Sciences ASSESSING THE DRUG ABILITY OF CHALCONES USING IN- SILICO TOOLS Department of Pharmaceutical Chemistry, Institute
Page109 IJPBS Volume 5 Issue 1 JAN-MAR 2015 109-114 Research Article Pharmaceutical Sciences ASSESSING THE DRUG ABILITY OF CHALCONES USING IN- SILICO TOOLS Department of Pharmaceutical Chemistry, Institute
Introduction to Chemoinformatics and Drug Discovery
 Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Advanced Medicinal Chemistry SLIDES B
 Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Plan. Day 2: Exercise on MHC molecules.
 Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Principles of Drug Design
 Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction
Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction
COMPUTER AIDED DRUG DESIGN (CADD) AND DEVELOPMENT METHODS
 AND DEVELOPMENT METHODS") COMPUTER AIDED DRUG DESIGN (CADD) AND DEVELOPMENT METHODS DRUG DEVELOPMENT Drug development is a challenging path Today, the causes of many diseases (rheumatoid arthritis, cancer, mental diseases, etc.)
COMPUTER AIDED DRUG DESIGN (CADD) AND DEVELOPMENT METHODS DRUG DEVELOPMENT Drug development is a challenging path Today, the causes of many diseases (rheumatoid arthritis, cancer, mental diseases, etc.)
Molecular docking, 3D-QSAR studies of indole hydrazone as Staphylococcus aureus pyruvate kinase inhibitor
 World Journal of Pharmaceutical Sciences ISSN (Print): 2321-3310; ISSN (Online): 2321-3086 Published by Atom and Cell Publishers All Rights Reserved Available online at: http://www.wjpsonline.org/ Original
World Journal of Pharmaceutical Sciences ISSN (Print): 2321-3310; ISSN (Online): 2321-3086 Published by Atom and Cell Publishers All Rights Reserved Available online at: http://www.wjpsonline.org/ Original
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Hydrogen Bonding & Molecular Design Peter
 Hydrogen Bonding & Molecular Design Peter Kenny(pwk.pub.2008@gmail.com) Hydrogen Bonding in Drug Discovery & Development Interactions between drug and water molecules (Solubility, distribution, permeability,
Hydrogen Bonding & Molecular Design Peter Kenny(pwk.pub.2008@gmail.com) Hydrogen Bonding in Drug Discovery & Development Interactions between drug and water molecules (Solubility, distribution, permeability,
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Research of New Class C beta-lactamase Inhibitor by Molecular Docking s Method
 Journal of Computational Methods in Molecular Design, 2018, 8 (1):26-32 ISSN : 2231-3176 CODEN (USA): JCMMD Research of New Class C beta-lactamase Inhibitor by Molecular Docking s Method Benhamoud Amina,
Journal of Computational Methods in Molecular Design, 2018, 8 (1):26-32 ISSN : 2231-3176 CODEN (USA): JCMMD Research of New Class C beta-lactamase Inhibitor by Molecular Docking s Method Benhamoud Amina,
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit
 October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
In Silico Investigation of Off-Target Effects
 PHARMA & LIFE SCIENCES WHITEPAPER In Silico Investigation of Off-Target Effects STREAMLINING IN SILICO PROFILING In silico techniques require exhaustive data and sophisticated, well-structured informatics
PHARMA & LIFE SCIENCES WHITEPAPER In Silico Investigation of Off-Target Effects STREAMLINING IN SILICO PROFILING In silico techniques require exhaustive data and sophisticated, well-structured informatics
TRAINING REAXYS MEDICINAL CHEMISTRY
 TRAINING REAXYS MEDICINAL CHEMISTRY 1 SITUATION: DRUG DISCOVERY Knowledge survey Therapeutic target Known ligands Generate chemistry ideas Chemistry Check chemical feasibility ELN DBs In-house Analyze
TRAINING REAXYS MEDICINAL CHEMISTRY 1 SITUATION: DRUG DISCOVERY Knowledge survey Therapeutic target Known ligands Generate chemistry ideas Chemistry Check chemical feasibility ELN DBs In-house Analyze
Molecular Modeling Study of Some Anthelmintic 2-phenyl Benzimidazole-1- Acetamides as β-tubulin Inhibitor
 Sawant et al : Molecular Modeling Study of Some Anthelmintic 2-phenyl Benzimidazole-1-Acetamides as -tubulin Inhibitor 1269 International Journal of Drug Design and Discovery Volume 5 Issue 1 January March
Sawant et al : Molecular Modeling Study of Some Anthelmintic 2-phenyl Benzimidazole-1-Acetamides as -tubulin Inhibitor 1269 International Journal of Drug Design and Discovery Volume 5 Issue 1 January March
11. IN SILICO DOCKING ANALYSIS
 11. IN SILICO DOCKING ANALYSIS 11.1 Molecular docking studies of major active constituents of ethanolic extract of GP The major active constituents are identified from the ethanolic extract of Glycosmis
11. IN SILICO DOCKING ANALYSIS 11.1 Molecular docking studies of major active constituents of ethanolic extract of GP The major active constituents are identified from the ethanolic extract of Glycosmis
Identifying Interaction Hot Spots with SuperStar
 Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
1. (18) Multiple choice questions. Please place your answer on the line preceding each question.
 Multiple choice questions. Please place your answer on the line preceding each question.") CEM 5720 ame KEY Exam 2 ctober 21, 2015 Read all questions carefully and attempt those questions you are sure of first. Remember to proof your work; art work carries as much importance as written responses.
CEM 5720 ame KEY Exam 2 ctober 21, 2015 Read all questions carefully and attempt those questions you are sure of first. Remember to proof your work; art work carries as much importance as written responses.
ADME SCREENING OF NOVEL 1,4-DIHYDROPYRIDINE DERIVATIVES
 ITERATIAL JURAL F RESEARC I PARMACY AD CEMISTRY Available online at www.ijrpc.com Research Article ADME SCREEIG F VEL 1,4-DIYDRPYRIDIE DERIVATIVES A. Asma samaunnisa 1*, CS. Venkataramana 1 and V Madhavan
ITERATIAL JURAL F RESEARC I PARMACY AD CEMISTRY Available online at www.ijrpc.com Research Article ADME SCREEIG F VEL 1,4-DIYDRPYRIDIE DERIVATIVES A. Asma samaunnisa 1*, CS. Venkataramana 1 and V Madhavan
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
FRAUNHOFER IME SCREENINGPORT
 FRAUNHOFER IME SCREENINGPORT Design of screening projects General remarks Introduction Screening is done to identify new chemical substances against molecular mechanisms of a disease It is a question of
FRAUNHOFER IME SCREENINGPORT Design of screening projects General remarks Introduction Screening is done to identify new chemical substances against molecular mechanisms of a disease It is a question of
DOCKING TUTORIAL. A. The docking Workflow
 2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
Principles of Drug Design
 (16:663:502) Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu For more current information, please check WebCT at https://webct.rutgers.edu
(16:663:502) Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu For more current information, please check WebCT at https://webct.rutgers.edu
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Enhancing Specificity in the Janus Kinases: A Study on the Thienopyridine. JAK2 Selective Mechanism Combined Molecular Dynamics Simulation
 Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is The Royal Society of Chemistry 2015 Supporting Information Enhancing Specificity in the Janus Kinases: A Study on the Thienopyridine
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is The Royal Society of Chemistry 2015 Supporting Information Enhancing Specificity in the Janus Kinases: A Study on the Thienopyridine
Ping-Chiang Lyu. Institute of Bioinformatics and Structural Biology, Department of Life Science, National Tsing Hua University.
 Pharmacophore-based Drug design Ping-Chiang Lyu Institute of Bioinformatics and Structural Biology, Department of Life Science, National Tsing Hua University 96/08/07 Outline Part I: Analysis The analytical
Pharmacophore-based Drug design Ping-Chiang Lyu Institute of Bioinformatics and Structural Biology, Department of Life Science, National Tsing Hua University 96/08/07 Outline Part I: Analysis The analytical
Alkane/water partition coefficients and hydrogen bonding. Peter Kenny
 Alkane/water partition coefficients and hydrogen bonding Peter Kenny (pwk.pub.2008@gmail.com) Neglect of hydrogen bond strength: A recurring theme in medicinal chemistry Rule of 5 Rule of 3 Scoring functions
Alkane/water partition coefficients and hydrogen bonding Peter Kenny (pwk.pub.2008@gmail.com) Neglect of hydrogen bond strength: A recurring theme in medicinal chemistry Rule of 5 Rule of 3 Scoring functions
Easier and Better Exploitation of PhysChem Properties in Medicinal Chemistry
 www.acdlabs.com Easier and Better Exploitation of PhysChem Properties in Medicinal Chemistry Dr. Sanjivanjit K. Bhal Advanced Chemistry Development, Inc. (ACD/Labs) Evolution of MedChem
www.acdlabs.com Easier and Better Exploitation of PhysChem Properties in Medicinal Chemistry Dr. Sanjivanjit K. Bhal Advanced Chemistry Development, Inc. (ACD/Labs) Evolution of MedChem
Introduction to medicinal chemisry
 2017 1 st edition Introduction to medicinal chemisry Med Chem I Mohammed Nooraldeen (PhD) كيمياء دوائية )1 ) محمد نورالدين محمود Introduction to medicinal chemistry Medicinal Chemistry Biology Medicine
2017 1 st edition Introduction to medicinal chemisry Med Chem I Mohammed Nooraldeen (PhD) كيمياء دوائية )1 ) محمد نورالدين محمود Introduction to medicinal chemistry Medicinal Chemistry Biology Medicine
Quantitative structure activity relationship (QSAR) of cardiac glycosides: the development of predictive in vitro cytotoxic activity model
 of cardiac glycosides: the development of predictive in vitro cytotoxic activity model") Available online at www.scholarsresearchlibrary.com Scholars Research Library Der Pharmacia Lettre, 2012, 4 (4):1246-1269 (http://scholarsresearchlibrary.com/archive.html) ISSN 0975-5071 USA CDEN: DPLEB4
Available online at www.scholarsresearchlibrary.com Scholars Research Library Der Pharmacia Lettre, 2012, 4 (4):1246-1269 (http://scholarsresearchlibrary.com/archive.html) ISSN 0975-5071 USA CDEN: DPLEB4
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Targeting protein-protein interactions: A hot topic in drug discovery
 Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
DOCKING OF THIOPURINE DERIVATIVES TO HUMAN SERUM ALBUMIN AND BINDING SITE ANALYSIS WITH MOLEGRO VIRTUAL DOCKER
 Acta Poloniae Pharmaceutica ñ Drug Research, Vol. 71 No. 2 pp. 343ñ349, 2014 ISSN 0001-6837 Polish Pharmaceutical Society SHORT COMMUNICATION DOCKING OF THIOPURINE DERIVATIVES TO HUMAN SERUM ALBUMIN AND
Acta Poloniae Pharmaceutica ñ Drug Research, Vol. 71 No. 2 pp. 343ñ349, 2014 ISSN 0001-6837 Polish Pharmaceutical Society SHORT COMMUNICATION DOCKING OF THIOPURINE DERIVATIVES TO HUMAN SERUM ALBUMIN AND
Computational Methods and Drug-Likeness. Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004
 Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
NMR study of complexes between low molecular mass inhibitors and the West Nile virus NS2B-NS3 protease
 University of Wollongong Research Online Faculty of Science - Papers (Archive) Faculty of Science, Medicine and Health 2009 NMR study of complexes between low molecular mass inhibitors and the West Nile
University of Wollongong Research Online Faculty of Science - Papers (Archive) Faculty of Science, Medicine and Health 2009 NMR study of complexes between low molecular mass inhibitors and the West Nile
Computational analysis of the activity of pongachalcone I against highly resistant bacteria Pseudomonas putida
 Computational analysis of the activity of pongachalcone I against highly resistant bacteria Pseudomonas putida Satya B. Paul 1, Sudip Choudhury 2,* 1 Department of Chemistry, Assam University, Silchar,
Computational analysis of the activity of pongachalcone I against highly resistant bacteria Pseudomonas putida Satya B. Paul 1, Sudip Choudhury 2,* 1 Department of Chemistry, Assam University, Silchar,
CHEM 4170 Problem Set #1
 CHEM 4170 Problem Set #1 0. Work problems 1-7 at the end of Chapter ne and problems 1, 3, 4, 5, 8, 10, 12, 17, 18, 19, 22, 24, and 25 at the end of Chapter Two and problem 1 at the end of Chapter Three
CHEM 4170 Problem Set #1 0. Work problems 1-7 at the end of Chapter ne and problems 1, 3, 4, 5, 8, 10, 12, 17, 18, 19, 22, 24, and 25 at the end of Chapter Two and problem 1 at the end of Chapter Three
Supplementary Information. Broad Spectrum Anti-Influenza Agents by Inhibiting Self- Association of Matrix Protein 1
 Supplementary Information Broad Spectrum Anti-Influenza Agents by Inhibiting Self- Association of Matrix Protein 1 Philip D. Mosier 1, Meng-Jung Chiang 2, Zhengshi Lin 2, Yamei Gao 2, Bashayer Althufairi
Supplementary Information Broad Spectrum Anti-Influenza Agents by Inhibiting Self- Association of Matrix Protein 1 Philip D. Mosier 1, Meng-Jung Chiang 2, Zhengshi Lin 2, Yamei Gao 2, Bashayer Althufairi
Quantitative structure activity relationship and drug design: A Review
 International Journal of Research in Biosciences Vol. 5 Issue 4, pp. (1-5), October 2016 Available online at http://www.ijrbs.in ISSN 2319-2844 Research Paper Quantitative structure activity relationship
International Journal of Research in Biosciences Vol. 5 Issue 4, pp. (1-5), October 2016 Available online at http://www.ijrbs.in ISSN 2319-2844 Research Paper Quantitative structure activity relationship
Design and Molecular Docking Studies of Some 1,3,4-Oxadiazole Derivatives
 Research Article Design and Molecular Docking Studies of Some 1,3,4-Oxadiazole Derivatives Dinesh Rishipathak *1, Prabhakar Shirodkar 2 1 Department of Pharmaceutical Chemistry, MET s Institute of Pharmacy,
Research Article Design and Molecular Docking Studies of Some 1,3,4-Oxadiazole Derivatives Dinesh Rishipathak *1, Prabhakar Shirodkar 2 1 Department of Pharmaceutical Chemistry, MET s Institute of Pharmacy,
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Plan. Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics.
 and its used in Chemoinformatics.") Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Synthetic organic compounds
 Synthetic organic compounds for research and drug discovery Compounds for TS Fragment libraries Target-focused libraries Chemical building blocks Custom synthesis Drug discovery services Contract research
Synthetic organic compounds for research and drug discovery Compounds for TS Fragment libraries Target-focused libraries Chemical building blocks Custom synthesis Drug discovery services Contract research
QSAR Study of Quinazoline Derivatives as Inhibitor of Epidermal Growth Factor Receptor-Tyrosine Kinase (EGFR-TK)
") 3rd International Conference on Computation for cience and Technology (ICCT-3) QAR tudy of Quinazoline Derivatives as Inhibitor of Epidermal Growth Factor Receptor-Tyrosine Kinase (EGFR-TK) La de Aman1*,
3rd International Conference on Computation for cience and Technology (ICCT-3) QAR tudy of Quinazoline Derivatives as Inhibitor of Epidermal Growth Factor Receptor-Tyrosine Kinase (EGFR-TK) La de Aman1*,
In Silico Design of New Drugs for Myeloid Leukemia Treatment
 In Silico Design of New Drugs for Myeloid Leukemia Treatment Washington Pereira and Ihosvany Camps Computational Modeling Laboratory LaModel Exact Science Institute ICEx Federal University of Alfenas -
In Silico Design of New Drugs for Myeloid Leukemia Treatment Washington Pereira and Ihosvany Camps Computational Modeling Laboratory LaModel Exact Science Institute ICEx Federal University of Alfenas -
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining
![Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining](/thumbs/91/107031261.jpg "Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining") Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
Quantum Mechanical Models of P450 Metabolism to Guide Optimization of Metabolic Stability
 Quantum Mechanical Models of P450 Metabolism to Guide Optimization of Metabolic Stability Optibrium Webinar 2015, June 17 2015 Jonathan Tyzack, Matthew Segall, Peter Hunt Optibrium, StarDrop, Auto-Modeller
Quantum Mechanical Models of P450 Metabolism to Guide Optimization of Metabolic Stability Optibrium Webinar 2015, June 17 2015 Jonathan Tyzack, Matthew Segall, Peter Hunt Optibrium, StarDrop, Auto-Modeller
Biological Macromolecules
 Introduction for Chem 493 Chemistry of Biological Macromolecules Dr. L. Luyt January 2008 Dr. L. Luyt Chem 493-2008 1 Biological macromolecules are the molecules of life allow for organization serve a
Introduction for Chem 493 Chemistry of Biological Macromolecules Dr. L. Luyt January 2008 Dr. L. Luyt Chem 493-2008 1 Biological macromolecules are the molecules of life allow for organization serve a
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
MOLECULAR DRUG TARGETS
 MOLECULAR DRUG TARGETS LEARNING OUTCOMES At the end of this session student shall be able to: List different types of druggable targets Describe forces involved in drug-receptor interactions Describe theories
MOLECULAR DRUG TARGETS LEARNING OUTCOMES At the end of this session student shall be able to: List different types of druggable targets Describe forces involved in drug-receptor interactions Describe theories
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites. J. Andrew Surface
 Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Life Sciences 1a Lecture Slides Set 10 Fall Prof. David R. Liu. Lecture Readings. Required: Lecture Notes McMurray p , O NH
 Life ciences 1a Lecture lides et 10 Fall 2006-2007 Prof. David R. Liu Lectures 17-18: The molecular basis of drug-protein binding: IV protease inhibitors 1. Drug development and its impact on IV-infected
Life ciences 1a Lecture lides et 10 Fall 2006-2007 Prof. David R. Liu Lectures 17-18: The molecular basis of drug-protein binding: IV protease inhibitors 1. Drug development and its impact on IV-infected
Virtual screening for drug discovery. Markus Lill Purdue University
 Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
Drug Discovery. Zainab Al Kharusi Office: 33-10
 Drug Discovery Zainab Al Kharusi Office: 33-10 Objectives: To understand the processes of Drug Development To be able to develop a plan for drug discovery Reference: Patrick G L. An Introduction to Medicinal
Drug Discovery Zainab Al Kharusi Office: 33-10 Objectives: To understand the processes of Drug Development To be able to develop a plan for drug discovery Reference: Patrick G L. An Introduction to Medicinal
Keywords: anti-coagulants, factor Xa, QSAR, Thrombosis. Introduction
 PostDoc Journal Vol. 2, No. 3, March 2014 Journal of Postdoctoral Research www.postdocjournal.com QSAR Study of Thiophene-Anthranilamides Based Factor Xa Direct Inhibitors Preetpal S. Sidhu Department
PostDoc Journal Vol. 2, No. 3, March 2014 Journal of Postdoctoral Research www.postdocjournal.com QSAR Study of Thiophene-Anthranilamides Based Factor Xa Direct Inhibitors Preetpal S. Sidhu Department
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Early Stages of Drug Discovery in the Pharmaceutical Industry
 Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
Viewing and Analyzing Proteins, Ligands and their Complexes 2
 2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
Different conformations of the drugs within the virtual library of FDA approved drugs will be generated.
 Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
QSAR of Microtubule Stabilizing Dictyostatins
 QSAR of Microtubule Stabilizing Dictyostatins Kia Montgomery BBSI 2007- University of Pittsburgh Department of Chemistry, Grambling State University Billy Day, Ph.D. Department of Pharmaceutical Sciences,
QSAR of Microtubule Stabilizing Dictyostatins Kia Montgomery BBSI 2007- University of Pittsburgh Department of Chemistry, Grambling State University Billy Day, Ph.D. Department of Pharmaceutical Sciences,
Build_model v User Guide
 Build_model v.2.0.1 User Guide MolTech Build_model User Guide 2008-2011 Molecular Technologies Ltd. www.moltech.ru Please send your comments and suggestions to contact@moltech.ru. Table of Contents Input
Build_model v.2.0.1 User Guide MolTech Build_model User Guide 2008-2011 Molecular Technologies Ltd. www.moltech.ru Please send your comments and suggestions to contact@moltech.ru. Table of Contents Input
CHEM J-9 June 2014
 CEM1611 2014-J-9 June 2014 Alanine (ala) and lysine (lys) are two amino acids with the structures given below as Fischer projections. The pk a values of the conjugate acid forms of the different functional
CEM1611 2014-J-9 June 2014 Alanine (ala) and lysine (lys) are two amino acids with the structures given below as Fischer projections. The pk a values of the conjugate acid forms of the different functional
Structure-Based Drug Discovery An Overview
 Structure-Based Drug Discovery An Overview Edited by Roderick E. Hubbard University of York, Heslington, York, UK and Vernalis (R&D) Ltd, Abington, Cambridge, UK RSC Publishing Contents Chapter 1 3D Structure
Structure-Based Drug Discovery An Overview Edited by Roderick E. Hubbard University of York, Heslington, York, UK and Vernalis (R&D) Ltd, Abington, Cambridge, UK RSC Publishing Contents Chapter 1 3D Structure
JCICS Major Research Areas
 JCICS Major Research Areas Chemical Information Text Searching Structure and Substructure Searching Databases Patents George W.A. Milne C571 Lecture Fall 2002 1 JCICS Major Research Areas Chemical Computation
JCICS Major Research Areas Chemical Information Text Searching Structure and Substructure Searching Databases Patents George W.A. Milne C571 Lecture Fall 2002 1 JCICS Major Research Areas Chemical Computation
Self-Organizing Molecular Field Analysis on a New Series of COX-2 Selective Inhibitors: 1,5-Diarylimidazoles
 Int. J. Mol. Sci. 2006, 7, 220-229 International Journal of Molecular Sciences ISSN 1422-0067 2006 by MDPI www.mdpi.org/ijms/ Self-Organizing Molecular Field Analysis on a New Series of COX-2 Selective
Int. J. Mol. Sci. 2006, 7, 220-229 International Journal of Molecular Sciences ISSN 1422-0067 2006 by MDPI www.mdpi.org/ijms/ Self-Organizing Molecular Field Analysis on a New Series of COX-2 Selective
Data Mining in the Chemical Industry. Overview of presentation
 Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Medicinal Chemistry/ CHEM 458/658 Chapter 3- SAR and QSAR
 Medicinal Chemistry/ CHEM 458/658 Chapter 3- SAR and QSAR Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Introduction Structure-Activity Relationship (SAR) - similar
Medicinal Chemistry/ CHEM 458/658 Chapter 3- SAR and QSAR Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Introduction Structure-Activity Relationship (SAR) - similar
Ligand-receptor interactions
 University of Silesia, Katowice, Poland 11 22 March 2013 Ligand-receptor interactions Dr. Pavel Polishchuk A.V. Bogatsky Physico-Chemical Institute of National Academy of Sciences of Ukraine Odessa, Ukraine
University of Silesia, Katowice, Poland 11 22 March 2013 Ligand-receptor interactions Dr. Pavel Polishchuk A.V. Bogatsky Physico-Chemical Institute of National Academy of Sciences of Ukraine Odessa, Ukraine
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
7.012 Problem Set 1. i) What are two main differences between prokaryotic cells and eukaryotic cells?
 What are two main differences between prokaryotic cells and eukaryotic cells?") ame 7.01 Problem Set 1 Section Question 1 a) What are the four major types of biological molecules discussed in lecture? Give one important function of each type of biological molecule in the cell? b)
ame 7.01 Problem Set 1 Section Question 1 a) What are the four major types of biological molecules discussed in lecture? Give one important function of each type of biological molecule in the cell? b)
Synthetic organic compounds
 Synthetic organic compounds for research and drug discovery chemicals Compounds for TS Fragment libraries Target-focused libraries Chemical building blocks Custom synthesis Drug discovery services Contract
Synthetic organic compounds for research and drug discovery chemicals Compounds for TS Fragment libraries Target-focused libraries Chemical building blocks Custom synthesis Drug discovery services Contract
IDENTIFICATION OF NOVEL INHIBITORS FOR MITOGEN-ACTIVATED PROTEIN KINASE KINASE 4 BY VIRTUAL SCREENING AND MOLECULAR DYNAMICS SIMULATION TECHNIQUES
 International Journal of Pharmacy and Pharmaceutical Sciences ISSN- 0975-1491 Vol 8, Issue 7, 2016 Original Article IDENTIFICATION OF NOVEL INHIBITORS FOR MITOGEN-ACTIVATED PROTEIN KINASE KINASE 4 BY VIRTUAL
International Journal of Pharmacy and Pharmaceutical Sciences ISSN- 0975-1491 Vol 8, Issue 7, 2016 Original Article IDENTIFICATION OF NOVEL INHIBITORS FOR MITOGEN-ACTIVATED PROTEIN KINASE KINASE 4 BY VIRTUAL
Ajeet, Jour. Harmo. Res. Pharm., 2013, 2(1), 45-53
, 45-53") Journal f Harmonized Research (JHR) Jounrnal f Harmonized Research in Pharmacy 2(1), 2013, 45-53 I 2321 0958 riginal Research Article DEVELPMET F QAR MDEL FR TUDYIG ULFAMIDE DERIVATIVE AGAIT CARBIC AHYDRAE
Journal f Harmonized Research (JHR) Jounrnal f Harmonized Research in Pharmacy 2(1), 2013, 45-53 I 2321 0958 riginal Research Article DEVELPMET F QAR MDEL FR TUDYIG ULFAMIDE DERIVATIVE AGAIT CARBIC AHYDRAE
Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,..
 3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
Comparison of log P/D with bio-mimetic properties
 Comparison of log P/D with bio-mimetic properties Klara Valko Bio-Mimetic Chromatography Consultancy for Successful Drug Discovery Solvation process First we need to create a cavity Solute molecule Requires
Comparison of log P/D with bio-mimetic properties Klara Valko Bio-Mimetic Chromatography Consultancy for Successful Drug Discovery Solvation process First we need to create a cavity Solute molecule Requires
Flexibility and Constraints in GOLD
 Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
Data Quality Issues That Can Impact Drug Discovery
 Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
NAME. EXAM I I. / 36 September 25, 2000 Biochemistry I II. / 26 BICH421/621 III. / 38 TOTAL /100
 EXAM I I. / 6 September 25, 2000 Biochemistry I II. / 26 BIH421/621 III. / 8 TOTAL /100 I. MULTIPLE HOIE (6 points) hoose the BEST answer to the question by circling the appropriate letter. 1. An amino
EXAM I I. / 6 September 25, 2000 Biochemistry I II. / 26 BIH421/621 III. / 8 TOTAL /100 I. MULTIPLE HOIE (6 points) hoose the BEST answer to the question by circling the appropriate letter. 1. An amino
Exam III. Please read through each question carefully, and make sure you provide all of the requested information.
 09-107 onors Chemistry ame Exam III Please read through each question carefully, and make sure you provide all of the requested information. 1. A series of octahedral metal compounds are made from 1 mol
09-107 onors Chemistry ame Exam III Please read through each question carefully, and make sure you provide all of the requested information. 1. A series of octahedral metal compounds are made from 1 mol
Introduction to medicinal chemisry. Mohammed Nooraldeen (PhD)
") Introduction to medicinal chemisry Med Chem I Mohammed Nooraldeen (PhD) كيمياء دوائية )1 ) محمد نورالدين محمود Introduction to medicinal chemistry Medicinal Chemistry Biology Medicine Biochemistry Physics
Introduction to medicinal chemisry Med Chem I Mohammed Nooraldeen (PhD) كيمياء دوائية )1 ) محمد نورالدين محمود Introduction to medicinal chemistry Medicinal Chemistry Biology Medicine Biochemistry Physics
Lead Optimization Studies on Novel Quinolones Derivatives as CYP-450 Inhibitor by using In-Silico Modulation
 World Journal of Pharmaceutical Sciences SS (Print): 2321-3310; SS (Online): 2321-3086 Available online at: http://www.wjpsonline.org/ Original Article Lead Optimization Studies on ovel Quinolones Derivatives
World Journal of Pharmaceutical Sciences SS (Print): 2321-3310; SS (Online): 2321-3086 Available online at: http://www.wjpsonline.org/ Original Article Lead Optimization Studies on ovel Quinolones Derivatives
Original Article Glycyrrhetinic acid derivatives on lung cancer: molecular docking, QSAR, ADME/T and In-Vitro analysis
 Int J Clin Exp Med 2016;9(2):3062-3068 www.ijcem.com /ISSN:1940-5901/IJCEM0015766 Original Article Glycyrrhetinic acid derivatives on lung cancer: molecular docking, QSAR, ADME/T and In-Vitro analysis
Int J Clin Exp Med 2016;9(2):3062-3068 www.ijcem.com /ISSN:1940-5901/IJCEM0015766 Original Article Glycyrrhetinic acid derivatives on lung cancer: molecular docking, QSAR, ADME/T and In-Vitro analysis
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Volume 12(2)
") Open access www.bioinformation.net Volume 12(2) Hypothesis Insights from molecular modeling, docking and simulation of imidazole nucleus containing chalcones with EGFR kinase domain for improved binding
Open access www.bioinformation.net Volume 12(2) Hypothesis Insights from molecular modeling, docking and simulation of imidazole nucleus containing chalcones with EGFR kinase domain for improved binding
Advanced Certificate in Principles in Protein Structure. You will be given a start time with your exam instructions
 BIRKBECK COLLEGE (University of London) Advanced Certificate in Principles in Protein Structure MSc Structural Molecular Biology Date: Thursday, 1st September 2011 Time: 3 hours You will be given a start
BIRKBECK COLLEGE (University of London) Advanced Certificate in Principles in Protein Structure MSc Structural Molecular Biology Date: Thursday, 1st September 2011 Time: 3 hours You will be given a start
Bioisosterism. A Rational Approach in Drug Design. 刘俊辉 Sept. 30th Sila-majantol 1b. lily-of-the-valley flowers but more terpineol-like odor
 Bioisosterism A Rational Approach in Drug Design 刘俊辉 Sept. 30th 2005 Why is Bioisosterism? Me C 2 C Me Majantol 1a strong fresh-floral aqueousaldehydic, lily-of-the-valley flowers odor Me C 2 Si Me Sila-majantol
Bioisosterism A Rational Approach in Drug Design 刘俊辉 Sept. 30th 2005 Why is Bioisosterism? Me C 2 C Me Majantol 1a strong fresh-floral aqueousaldehydic, lily-of-the-valley flowers odor Me C 2 Si Me Sila-majantol
Supporting Information (Part II) for ACS Combinatorial Science
 for ACS Combinatorial Science") Supporting Information (Part II) for ACS Combinatorial Science Application of 6,7Indole Aryne Cycloaddition and Pd(0)Catalyzed SuzukiMiyaura and BuchwaldHartwig CrossCoupling Reactions for the Preparation
Supporting Information (Part II) for ACS Combinatorial Science Application of 6,7Indole Aryne Cycloaddition and Pd(0)Catalyzed SuzukiMiyaura and BuchwaldHartwig CrossCoupling Reactions for the Preparation
Packing of Secondary Structures
 7.88 Lecture Notes - 4 7.24/7.88J/5.48J The Protein Folding and Human Disease Professor Gossard Retrieving, Viewing Protein Structures from the Protein Data Base Helix helix packing Packing of Secondary
7.88 Lecture Notes - 4 7.24/7.88J/5.48J The Protein Folding and Human Disease Professor Gossard Retrieving, Viewing Protein Structures from the Protein Data Base Helix helix packing Packing of Secondary
Nonlinear QSAR and 3D QSAR
 onlinear QSAR and 3D QSAR Hugo Kubinyi Germany E-Mail kubinyi@t-online.de HomePage www.kubinyi.de onlinear Lipophilicity-Activity Relationships drug receptor Possible Reasons for onlinear Lipophilicity-Activity
onlinear QSAR and 3D QSAR Hugo Kubinyi Germany E-Mail kubinyi@t-online.de HomePage www.kubinyi.de onlinear Lipophilicity-Activity Relationships drug receptor Possible Reasons for onlinear Lipophilicity-Activity
Molecular docking studies on mycobacterial tuberculosis-6-oxopurine phosphoribosyltransferase EMRB with pyrimidine derivatives
 Journal of Computational Methods in Molecular Design, 2015, 5 (4):120-128 Scholars Research Library (http://scholarsresearchlibrary.com/archive.html) ISS : 2231-3176 CDE (USA): JCMMDA Molecular docking
Journal of Computational Methods in Molecular Design, 2015, 5 (4):120-128 Scholars Research Library (http://scholarsresearchlibrary.com/archive.html) ISS : 2231-3176 CDE (USA): JCMMDA Molecular docking
Course Specification Medicinal Chemistry-1
 Quality Assurance Unit Faculty of Pharmacy-Assiut University Course Specification Medicinal Chemistry-1 Programme(s) on which the course is given: B. Sc. (Pharmaceutical science) Major or Minor element
Quality Assurance Unit Faculty of Pharmacy-Assiut University Course Specification Medicinal Chemistry-1 Programme(s) on which the course is given: B. Sc. (Pharmaceutical science) Major or Minor element
Solutions and Non-Covalent Binding Forces
 Chapter 3 Solutions and Non-Covalent Binding Forces 3.1 Solvent and solution properties Molecules stick together using the following forces: dipole-dipole, dipole-induced dipole, hydrogen bond, van der
Chapter 3 Solutions and Non-Covalent Binding Forces 3.1 Solvent and solution properties Molecules stick together using the following forces: dipole-dipole, dipole-induced dipole, hydrogen bond, van der
A primer on pharmacology pharmacodynamics
 A primer on pharmacology pharmacodynamics Drug binding & effect Universidade do Algarve Faro 2017 by Ferdi Engels, Ph.D. 1 Pharmacodynamics Relation with pharmacokinetics? dosage plasma concentration site
A primer on pharmacology pharmacodynamics Drug binding & effect Universidade do Algarve Faro 2017 by Ferdi Engels, Ph.D. 1 Pharmacodynamics Relation with pharmacokinetics? dosage plasma concentration site
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific