Predictive Molecular Simulation for Drug Discovery
|
|
|
- Meredith Lloyd
- 6 years ago
- Views:
Transcription

1 Predictive Molecular Simulation for Drug Discovery 생명과학 의약연구소의약설계팀이승주
2 Outline Challenging problems in drug discovery Binding affinity calculation New paradigm for scientific computing

3 Challenging problems in Drug Discovery Prediction of Binding Affinity
4 Challenges for molecular simulation in Drug Discovery: Prediction Prediction ( 미래형 ) Use faster and cheaper methods to predict outcomes of more expensive methods 기상청 : 내일의날씨 내일폭등할주식 Retrodiction ( 과거형 ) Explain what happened Unavailable until after the fact El Nino 현상분석 어제폭등한주식
 Identification ->")

5 Cycle of Drug Discovery Lead(hit) Identification -> Lead Optimization -> Preclinical Development 10 4 ~ 10 5 개 ~10 mm ~10 nm
6 Lead identification is feasible Find starting material IC 50 < 10 mm virtual HTS docking or QSAR Linux cluster 6 x 10 5 cpd in days CPU: ~minute per compound comparable to HTS ~mm activity 까지는찾을수있다. ~mm 이하는변별력이없다 Docking score vs Experiment Need predictive strength in the ~nm range for lead optimization Reducing hit to lead time is important: 6months ~ 9months Org. Biomol. Chem., 2004, 2, (22),
7 Lead optimization is difficult: what is necessary? Hardware Linux cluster CPU scavenging Software accurate force field fast molecular dynamics code Theory Free energy calculation methodology Linear Interaction Energy Free energy perturbation Bennett s acceptance ratio JCP,123, , cpu days



8 High Throughput Molecular Dynamics at LG Life Sciences Docking Glide Protein Modeling BACE inhibitor 치매치료제 Partial Charge Force Field GAFF AMBER99 TIP3P Molecular Dynamics Gromacs on PC United! Linux cluster Free Energy Calculation Linear Interaction Energy

9 Docking -> MD Docking mode might be wrong, but DG might be similar Multiple binding modes observed in xtal structures Multiple binding modes can be Boltzmann weighted Camacho and Vajda PNAS 2001,10636

10 Protein modeling: BACE Protonation state of important Asp residues not resolved 8 possible states Protonation state may depend on ligand
11 Free Energy Calculation Linear Interaction Energy(LIE) input Protein+drug+water의 MD 계산결과 drug+water 의 MD 계산결과 output Protein+ drug 의 free energy 계산 G binding ( V ( V elec ligand protein vdw ligand protein V V elec ligand solvent vdw ligand solvent ) ) J Aqvist et al Acc. Chem. Res, 2002,35,358 Van Lipzig, J Med Chem, 2004,47,1018

")


")

12 CPU LG Life Sciences 사내 Condor high throughput computing 시스템구축 PC United 프로젝트 (2006 년 10 월개시 ) client 설치자동화 유휴 CPU 활용 Configuration 1 central server ( Linux PC ) ~100 Microsoft Windows XP clients 30 CPU개 cluster와비슷 Condor queueing system Wisconsin 대학개발 ( 무료 ) Windows, Linux, Unix 지원 Windows에서 compile가능한 code 실행가능 (cygwin) 현재 Gromacs 실행
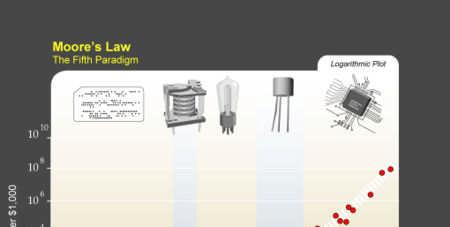
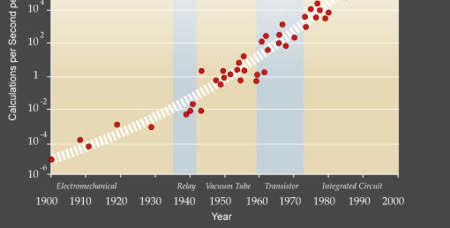
13


14 The Sixth Paradigm? 1997: $30,000/GFLOPS Linux cluster 2000: $640/GFLOPS, Linux cluster 2003: $82/GFLOPS Linux cluster 2005: $2.60/GFLOPS Xbox : $1/GFLOPS ATI graphics card


15 Supercomputing s Next Revolution GPU based computing Supercomputing s Next Revolution 11/9/2006, Wired News
 2.")
 1 TFlop Graphics Card (ATI, Jan 2006) Radeon X1900, x1950")
16 Supercomputing s Next Revolution GPU based computing Game Console Computing Sony PS3 runs MD (Folding@Home) 2.18 TFlops total /PS3 ~100GFlops / PS3 for Gromacs Xbox 360 ~1TFlops Graphics Card Computing Gromacs runs on GPU (Folding@Home) 1 TFlop Graphics Card (ATI, Jan 2006) Radeon X1900, x , first Tflop computer ASCI Red 9,200 Pentium II chips Gromacs runs 20~40 times faster
 support from games to genes GPU version of MD Gromacs http://graphics.stanford.")
17 GPU based Computing Environment C programming environment BrooksGPU (Stanford) Nvidia CUDA ATI (AMD) support from games to genes GPU version of MD Gromacs

18 Molecular Simulation Based drug toxicity prediction Drug cytochrome P450 binding affinity drug-drug interaction 예측가능 최근 X-ray 구조발표보다정확하게예측가능함 Nature 424, ,2003 J. Med. Chem., 47 (22), 5340, 2004 Homology model + docking


19 Molecular Simulation Based drug toxicity prediction Drug HERG K+ channel binding affinity 예측 HERG 를 inhibit 하면 cardiovascular toxicity 보임 Homology model+docking PNAS 2000 vol cited 257 times 독성예측은 hot topic user base 넓다 web based service 가능 commercialize 가능 ADMET in silico modelling: towards prediction paradise? Nature Review Drug Discovery, 2003, 2: 192
20 Summary Challenging problems in drug discovery Many challenging problems for molecular simulation Accurate prediction of binding affinity < mm Revolution in speed: stream computing Application for molecular simulation Cytochrome P450 inhibition HERG channel inhibition
21 Thanks to LG 생명과학 Dr. 임동철 Dr. 고종성 Kaist 이장환 ICU Computational Systems Biology 선충현, 김진기
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors
 MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
Early Stages of Drug Discovery in the Pharmaceutical Industry
 Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today
 est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
Molecular dynamics simulation. CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror
 Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
5.1. Hardwares, Softwares and Web server used in Molecular modeling
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
Virtual Screening: How Are We Doing?
 Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Adaptive Heterogeneous Computing with OpenCL: Harnessing hundreds of GPUs and CPUs
 Adaptive Heterogeneous Computing with OpenCL: Harnessing hundreds of GPUs and CPUs Simon McIntosh-Smith simonm@cs.bris.ac.uk Head of Microelectronics Research University of Bristol, UK 1 ! Collaborators
Adaptive Heterogeneous Computing with OpenCL: Harnessing hundreds of GPUs and CPUs Simon McIntosh-Smith simonm@cs.bris.ac.uk Head of Microelectronics Research University of Bristol, UK 1 ! Collaborators
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
FRAGMENT SCREENING IN LEAD DISCOVERY BY WEAK AFFINITY CHROMATOGRAPHY (WAC )
") FRAGMENT SCREENING IN LEAD DISCOVERY BY WEAK AFFINITY CHROMATOGRAPHY (WAC ) SARomics Biostructures AB & Red Glead Discovery AB Medicon Village, Lund, Sweden Fragment-based lead discovery The basic idea:
FRAGMENT SCREENING IN LEAD DISCOVERY BY WEAK AFFINITY CHROMATOGRAPHY (WAC ) SARomics Biostructures AB & Red Glead Discovery AB Medicon Village, Lund, Sweden Fragment-based lead discovery The basic idea:
MM-PBSA Validation Study. Trent E. Balius Department of Applied Mathematics and Statistics AMS
 MM-PBSA Validation Study Trent. Balius Department of Applied Mathematics and Statistics AMS 535 11-26-2008 Overview MM-PBSA Introduction MD ensembles one snap-shots relaxed structures nrichment Computational
MM-PBSA Validation Study Trent. Balius Department of Applied Mathematics and Statistics AMS 535 11-26-2008 Overview MM-PBSA Introduction MD ensembles one snap-shots relaxed structures nrichment Computational
Hit Finding and Optimization Using BLAZE & FORGE
 Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Cross Discipline Analysis made possible with Data Pipelining. J.R. Tozer SciTegic
 Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
MD Simulation in Pose Refinement and Scoring Using AMBER Workflows
 MD Simulation in Pose Refinement and Scoring Using AMBER Workflows Yuan Hu (On behalf of Merck D3R Team) D3R Grand Challenge 2 Webinar Department of Chemistry, Modeling & Informatics Merck Research Laboratories,
MD Simulation in Pose Refinement and Scoring Using AMBER Workflows Yuan Hu (On behalf of Merck D3R Team) D3R Grand Challenge 2 Webinar Department of Chemistry, Modeling & Informatics Merck Research Laboratories,
Alchemical free energy calculations in OpenMM
 Alchemical free energy calculations in OpenMM Lee-Ping Wang Stanford Department of Chemistry OpenMM Workshop, Stanford University September 7, 2012 Special thanks to: John Chodera, Morgan Lawrenz Outline
Alchemical free energy calculations in OpenMM Lee-Ping Wang Stanford Department of Chemistry OpenMM Workshop, Stanford University September 7, 2012 Special thanks to: John Chodera, Morgan Lawrenz Outline
Advanced Medicinal Chemistry SLIDES B
 Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Principles of Drug Design
 Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction
Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction
BUDE. A General Purpose Molecular Docking Program Using OpenCL. Richard B Sessions
 BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,..
 3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
Building innovative drug discovery alliances. Just in KNIME: Successful Process Driven Drug Discovery
 Building innovative drug discovery alliances Just in KIME: Successful Process Driven Drug Discovery Berlin KIME Spring Summit, Feb 2016 Research Informatics @ Evotec Evotec s worldwide operations 2 Pharmaceuticals
Building innovative drug discovery alliances Just in KIME: Successful Process Driven Drug Discovery Berlin KIME Spring Summit, Feb 2016 Research Informatics @ Evotec Evotec s worldwide operations 2 Pharmaceuticals
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Statistical Clustering of Vesicle Patterns Practical Aspects of the Analysis of Large Datasets with R
 Statistical Clustering of Vesicle Patterns Mirko Birbaumer Rmetrics Workshop 3th July 2008 1 / 23 Statistical Clustering of Vesicle Patterns Practical Aspects of the Analysis of Large Datasets with R Mirko
Statistical Clustering of Vesicle Patterns Mirko Birbaumer Rmetrics Workshop 3th July 2008 1 / 23 Statistical Clustering of Vesicle Patterns Practical Aspects of the Analysis of Large Datasets with R Mirko
MURCIA: Fast parallel solvent accessible surface area calculation on GPUs and application to drug discovery and molecular visualization
 MURCIA: Fast parallel solvent accessible surface area calculation on GPUs and application to drug discovery and molecular visualization Eduardo J. Cepas Quiñonero Horacio Pérez-Sánchez Wolfgang Wenzel
MURCIA: Fast parallel solvent accessible surface area calculation on GPUs and application to drug discovery and molecular visualization Eduardo J. Cepas Quiñonero Horacio Pérez-Sánchez Wolfgang Wenzel
VMD: Visual Molecular Dynamics. Our Microscope is Made of...
 VMD: Visual Molecular Dynamics Computational Microscope / Tool to Think amino acid tyrosine enzymatic control BPTI VMD tutorial traficking Ubiquitin case study http://www.ks.uiuc.edu/training/casestudies/
VMD: Visual Molecular Dynamics Computational Microscope / Tool to Think amino acid tyrosine enzymatic control BPTI VMD tutorial traficking Ubiquitin case study http://www.ks.uiuc.edu/training/casestudies/
FRAUNHOFER IME SCREENINGPORT
 FRAUNHOFER IME SCREENINGPORT Design of screening projects General remarks Introduction Screening is done to identify new chemical substances against molecular mechanisms of a disease It is a question of
FRAUNHOFER IME SCREENINGPORT Design of screening projects General remarks Introduction Screening is done to identify new chemical substances against molecular mechanisms of a disease It is a question of
Cheminformatics platform for drug discovery application
 EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
NIH Center for Macromolecular Modeling and Bioinformatics Developer of VMD and NAMD. Beckman Institute
 NIH Center for Macromolecular Modeling and Bioinformatics Developer of VMD and NAMD 5 faculty members (2 physics, 1 chemistry, 1 biochemistry, 1 computer science); 8 developers; 1 system admin; 15 post
NIH Center for Macromolecular Modeling and Bioinformatics Developer of VMD and NAMD 5 faculty members (2 physics, 1 chemistry, 1 biochemistry, 1 computer science); 8 developers; 1 system admin; 15 post
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery
 21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
Integrated Cheminformatics to Guide Drug Discovery
 Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Structure-based maximal affinity model predicts small-molecule druggability
 Structure-based maximal affinity model predicts small-molecule druggability Alan Cheng alan.cheng@amgen.com IMA Workshop (Jan 17, 2008) Druggability prediction Introduction Affinity model Some results
Structure-based maximal affinity model predicts small-molecule druggability Alan Cheng alan.cheng@amgen.com IMA Workshop (Jan 17, 2008) Druggability prediction Introduction Affinity model Some results
NIH Center for Macromolecular Modeling and Bioinformatics Developer of VMD and NAMD. Beckman Institute
 NIH Center for Macromolecular Modeling and Bioinformatics Developer of VMD and NAMD 5 faculty members (2 physics, 1 chemistry, 1 biochemistry, 1 computer science); 8 developers; 1 system admin; 15 post
NIH Center for Macromolecular Modeling and Bioinformatics Developer of VMD and NAMD 5 faculty members (2 physics, 1 chemistry, 1 biochemistry, 1 computer science); 8 developers; 1 system admin; 15 post
Introduction to Chemoinformatics and Drug Discovery
 Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Mechanisms for exploiting heterogeneous computing: Harnessing hundreds of GPUs and CPUs
 Mechanisms for exploiting heterogeneous computing: Harnessing hundreds of GPUs and CPUs Simon McIntosh-Smith simonm@cs.bris.ac.uk Head of Microelectronics Research University of Bristol, UK 1 ! Collaborators
Mechanisms for exploiting heterogeneous computing: Harnessing hundreds of GPUs and CPUs Simon McIntosh-Smith simonm@cs.bris.ac.uk Head of Microelectronics Research University of Bristol, UK 1 ! Collaborators
Implementation of novel tools to facilitate fragment-based drug discovery by NMR:
 Implementation of novel tools to facilitate fragment-based drug discovery by NMR: Automated analysis of large sets of ligand-observed NMR binding data and 19 F methods Andreas Lingel Global Discovery Chemistry
Implementation of novel tools to facilitate fragment-based drug discovery by NMR: Automated analysis of large sets of ligand-observed NMR binding data and 19 F methods Andreas Lingel Global Discovery Chemistry
Cheminformatics Role in Pharmaceutical Industry. Randal Chen Ph.D. Abbott Laboratories Aug. 23, 2004 ACS
 Cheminformatics Role in Pharmaceutical Industry Randal Chen Ph.D. Abbott Laboratories Aug. 23, 2004 ACS Agenda The big picture for pharmaceutical industry Current technological/scientific issues Types
Cheminformatics Role in Pharmaceutical Industry Randal Chen Ph.D. Abbott Laboratories Aug. 23, 2004 ACS Agenda The big picture for pharmaceutical industry Current technological/scientific issues Types
Structure based drug design and LIE models for GPCRs
 Structure based drug design and LIE models for GPCRs Peter Kolb kolb@docking.org Shoichet Lab ACS 237 th National Meeting, March 24, 2009 p.1/26 [Acknowledgements] Brian Shoichet John Irwin Mike Keiser
Structure based drug design and LIE models for GPCRs Peter Kolb kolb@docking.org Shoichet Lab ACS 237 th National Meeting, March 24, 2009 p.1/26 [Acknowledgements] Brian Shoichet John Irwin Mike Keiser
Principles of Drug Design
 (16:663:502) Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu For more current information, please check WebCT at https://webct.rutgers.edu
(16:663:502) Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu For more current information, please check WebCT at https://webct.rutgers.edu
Parallelization of Molecular Dynamics (with focus on Gromacs) SeSE 2014 p.1/29
 SeSE 2014 p.1/29") Parallelization of Molecular Dynamics (with focus on Gromacs) SeSE 2014 p.1/29 Outline A few words on MD applications and the GROMACS package The main work in an MD simulation Parallelization Stream computing
Parallelization of Molecular Dynamics (with focus on Gromacs) SeSE 2014 p.1/29 Outline A few words on MD applications and the GROMACS package The main work in an MD simulation Parallelization Stream computing
Molecular Dynamics. Molecules in motion
 Molecular Dynamics Molecules in motion 1 Molecules in mo1on Molecules are not sta1c, but move all the 1me Source: h9p://en.wikipedia.org/wiki/kine1c_theory 2 Gasses, liquids and solids Gasses, liquids
Molecular Dynamics Molecules in motion 1 Molecules in mo1on Molecules are not sta1c, but move all the 1me Source: h9p://en.wikipedia.org/wiki/kine1c_theory 2 Gasses, liquids and solids Gasses, liquids
Introduction to FBDD Fragment screening methods and library design
 Introduction to FBDD Fragment screening methods and library design Samantha Hughes, PhD Fragments 2013 RSC BMCS Workshop 3 rd March 2013 Copyright 2013 Galapagos NV Why fragment screening methods? Guess
Introduction to FBDD Fragment screening methods and library design Samantha Hughes, PhD Fragments 2013 RSC BMCS Workshop 3 rd March 2013 Copyright 2013 Galapagos NV Why fragment screening methods? Guess
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
High Throughput In-Silico Screening Against Flexible Protein Receptors
 John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
GC and CELPP: Workflows and Insights
 GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
Lecture for Molekylär bioinformatik X3 Feb Computational Chemistry in Drug Discovery. Mats Kihlén. Head of Research Informatics.
 Lecture for Molekylär bioinformatik X3 Feb 24 2004 Computational Chemistry in Drug Discovery Mats Kihlén Head of Research Informatics Biovitrum AB verview» The role of computational chemistry» Basic concepts
Lecture for Molekylär bioinformatik X3 Feb 24 2004 Computational Chemistry in Drug Discovery Mats Kihlén Head of Research Informatics Biovitrum AB verview» The role of computational chemistry» Basic concepts
OpenDiscovery: Automated Docking of Ligands to Proteins and Molecular Simulation
 OpenDiscovery: Automated Docking of Ligands to Proteins and Molecular Simulation Gareth Price Computational MiniProject OpenDiscovery Aims + Achievements Produce a high-throughput protocol to screen a
OpenDiscovery: Automated Docking of Ligands to Proteins and Molecular Simulation Gareth Price Computational MiniProject OpenDiscovery Aims + Achievements Produce a high-throughput protocol to screen a
Introduction to Protein Structures - Molecular Graphics Tool
 Lecture 1a Introduction to Protein Structures - Molecular Graphics Tool amino acid tyrosine LH2 energetics traficking Ubiquitin enzymatic control BPTI Highlights of the VMD Molecular Graphics Program >
Lecture 1a Introduction to Protein Structures - Molecular Graphics Tool amino acid tyrosine LH2 energetics traficking Ubiquitin enzymatic control BPTI Highlights of the VMD Molecular Graphics Program >
Ignasi Belda, PhD CEO. HPC Advisory Council Spain Conference 2015
 Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Advanced Molecular Dynamics
 Advanced Molecular Dynamics Introduction May 2, 2017 Who am I? I am an associate professor at Theoretical Physics Topics I work on: Algorithms for (parallel) molecular simulations including GPU acceleration
Advanced Molecular Dynamics Introduction May 2, 2017 Who am I? I am an associate professor at Theoretical Physics Topics I work on: Algorithms for (parallel) molecular simulations including GPU acceleration
Welcome to MCS 572. content and organization expectations of the course. definition and classification
 Welcome to MCS 572 1 About the Course content and organization expectations of the course 2 Supercomputing definition and classification 3 Measuring Performance speedup and efficiency Amdahl s Law Gustafson
Welcome to MCS 572 1 About the Course content and organization expectations of the course 2 Supercomputing definition and classification 3 Measuring Performance speedup and efficiency Amdahl s Law Gustafson
*Corresponding Author *K. F.: *T. H.:
 Theoretical Analysis of Activity Cliffs among Benzofuranone Class Pim1 Inhibitors Using the Fragment Molecular Orbital Method with Molecular Mechanics Poisson-Boltzmann Surface Area (FMO+MM-PBSA) Approach
Theoretical Analysis of Activity Cliffs among Benzofuranone Class Pim1 Inhibitors Using the Fragment Molecular Orbital Method with Molecular Mechanics Poisson-Boltzmann Surface Area (FMO+MM-PBSA) Approach
TRAINING REAXYS MEDICINAL CHEMISTRY
 TRAINING REAXYS MEDICINAL CHEMISTRY 1 SITUATION: DRUG DISCOVERY Knowledge survey Therapeutic target Known ligands Generate chemistry ideas Chemistry Check chemical feasibility ELN DBs In-house Analyze
TRAINING REAXYS MEDICINAL CHEMISTRY 1 SITUATION: DRUG DISCOVERY Knowledge survey Therapeutic target Known ligands Generate chemistry ideas Chemistry Check chemical feasibility ELN DBs In-house Analyze
MSc Drug Design. Module Structure: (15 credits each) Lectures and Tutorials Assessment: 50% coursework, 50% unseen examination.
 Lectures and Tutorials Assessment: 50% coursework, 50% unseen examination.") Module Structure: (15 credits each) Lectures and Assessment: 50% coursework, 50% unseen examination. Module Title Module 1: Bioinformatics and structural biology as applied to drug design MEDC0075 In the
Module Structure: (15 credits each) Lectures and Assessment: 50% coursework, 50% unseen examination. Module Title Module 1: Bioinformatics and structural biology as applied to drug design MEDC0075 In the
History of Scientific Computing!
 History of Scientific Computing! Topics to be addressed: Growth of compu5ng power Beginnings of Computa5onal Chemistry History of modern opera5ng system for scien5fic compu5ng: UNIX Current compu5ng power
History of Scientific Computing! Topics to be addressed: Growth of compu5ng power Beginnings of Computa5onal Chemistry History of modern opera5ng system for scien5fic compu5ng: UNIX Current compu5ng power
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design
 Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Course Plan (Syllabus): Drug Design and Discovery
: Drug Design and Discovery") Course Plan (Syllabus): Drug Design and Discovery (A) Course Identification and General Information Course Number & Code PPC 515 Course Title Drug Design and Discovery (Elective course) Program (s) in
Course Plan (Syllabus): Drug Design and Discovery (A) Course Identification and General Information Course Number & Code PPC 515 Course Title Drug Design and Discovery (Elective course) Program (s) in
Capturing Chemistry. What you see is what you get In the world of mechanism and chemical transformations
 Capturing Chemistry What you see is what you get In the world of mechanism and chemical transformations Dr. Stephan Schürer ead of Intl. Sci. Content Libraria, Inc. sschurer@libraria.com Distribution of
Capturing Chemistry What you see is what you get In the world of mechanism and chemical transformations Dr. Stephan Schürer ead of Intl. Sci. Content Libraria, Inc. sschurer@libraria.com Distribution of
Large Scale FEP on Congeneric Ligand Series Have Practical Free Energy Calculations arrived at Last?
 Large Scale FEP on Congeneric Ligand Series Have Practical Free Energy Calculations arrived at Last? Thomas Steinbrecher, Teng Lin, Lingle Wang, Goran Krilov, Robert Abel, Woody Sherman, Richard Friesner
Large Scale FEP on Congeneric Ligand Series Have Practical Free Energy Calculations arrived at Last? Thomas Steinbrecher, Teng Lin, Lingle Wang, Goran Krilov, Robert Abel, Woody Sherman, Richard Friesner
György M. Keserű H2020 FRAGNET Network Hungarian Academy of Sciences
 Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Chemoinformatics and information management. Peter Willett, University of Sheffield, UK
 Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
The Changing Requirements for Informatics Systems During the Growth of a Collaborative Drug Discovery Service Company. Sally Rose BioFocus plc
 The Changing Requirements for Informatics Systems During the Growth of a Collaborative Drug Discovery Service Company Sally Rose BioFocus plc Overview History of BioFocus and acquisition of CDD Biological
The Changing Requirements for Informatics Systems During the Growth of a Collaborative Drug Discovery Service Company Sally Rose BioFocus plc Overview History of BioFocus and acquisition of CDD Biological
Isothermal Titration Calorimetry in Drug Discovery. Geoff Holdgate Structure & Biophysics, Discovery Sciences, AstraZeneca October 2017
 Isothermal Titration Calorimetry in Drug Discovery Geoff Holdgate Structure & Biophysics, Discovery Sciences, AstraZeneca October 217 Introduction Introduction to ITC Strengths / weaknesses & what is required
Isothermal Titration Calorimetry in Drug Discovery Geoff Holdgate Structure & Biophysics, Discovery Sciences, AstraZeneca October 217 Introduction Introduction to ITC Strengths / weaknesses & what is required
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Challenges, Applications, and Recent Advances of Protein-Ligand Docking in Structure-Based Drug Design
 Molecules 2014, 19, 10150-10176; doi:10.3390/molecules190710150 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Challenges, Applications, and Recent Advances of Protein-Ligand
Molecules 2014, 19, 10150-10176; doi:10.3390/molecules190710150 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Challenges, Applications, and Recent Advances of Protein-Ligand
Quantitative structure activity relationship and drug design: A Review
 International Journal of Research in Biosciences Vol. 5 Issue 4, pp. (1-5), October 2016 Available online at http://www.ijrbs.in ISSN 2319-2844 Research Paper Quantitative structure activity relationship
International Journal of Research in Biosciences Vol. 5 Issue 4, pp. (1-5), October 2016 Available online at http://www.ijrbs.in ISSN 2319-2844 Research Paper Quantitative structure activity relationship
Haralambos Tzoupis Georgios Leonis Serdar Durdagi Varnavas Mouchlis Thomas Mavromoustakos Manthos G. Papadopoulos
 J Comput Aided Mol Des (2011) 25:959 976 DI 10.1007/s10822-011-9475-4 Binding of novel fullerene inhibitors to HIV-1 protease: insight through molecular dynamics and molecular mechanics Poisson Boltzmann
J Comput Aided Mol Des (2011) 25:959 976 DI 10.1007/s10822-011-9475-4 Binding of novel fullerene inhibitors to HIV-1 protease: insight through molecular dynamics and molecular mechanics Poisson Boltzmann
Computationally Efficient Analysis of Large Array FTIR Data In Chemical Reaction Studies Using Distributed Computing Strategy
 575f Computationally Efficient Analysis of Large Array FTIR Data In Chemical Reaction Studies Using Distributed Computing Strategy Ms Suyun Ong, Dr. Wee Chew, * Dr. Marc Garland Institute of Chemical and
575f Computationally Efficient Analysis of Large Array FTIR Data In Chemical Reaction Studies Using Distributed Computing Strategy Ms Suyun Ong, Dr. Wee Chew, * Dr. Marc Garland Institute of Chemical and
Improving Protein Function Prediction with Molecular Dynamics Simulations. Dariya Glazer Russ Altman
 Improving Protein Function Prediction with Molecular Dynamics Simulations Dariya Glazer Russ Altman Motivation Sometimes the 3D structure doesn t score well for a known function. The experimental structure
Improving Protein Function Prediction with Molecular Dynamics Simulations Dariya Glazer Russ Altman Motivation Sometimes the 3D structure doesn t score well for a known function. The experimental structure
Direct Self-Consistent Field Computations on GPU Clusters
 Direct Self-Consistent Field Computations on GPU Clusters Guochun Shi, Volodymyr Kindratenko National Center for Supercomputing Applications University of Illinois at UrbanaChampaign Ivan Ufimtsev, Todd
Direct Self-Consistent Field Computations on GPU Clusters Guochun Shi, Volodymyr Kindratenko National Center for Supercomputing Applications University of Illinois at UrbanaChampaign Ivan Ufimtsev, Todd
Fundamentals of Computational Science
 Fundamentals of Computational Science Dr. Hyrum D. Carroll August 23, 2016 Introductions Each student: Name Undergraduate school & major Masters & major Previous research (if any) Why Computational Science
Fundamentals of Computational Science Dr. Hyrum D. Carroll August 23, 2016 Introductions Each student: Name Undergraduate school & major Masters & major Previous research (if any) Why Computational Science
Investigation of physiochemical interactions in
 Investigation of physiochemical interactions in Bulk and interfacial water Aqueous salt solutions (new endeavor) Polypeptides exhibiting a helix-coil transition Aqueous globular proteins Protein-solvent
Investigation of physiochemical interactions in Bulk and interfacial water Aqueous salt solutions (new endeavor) Polypeptides exhibiting a helix-coil transition Aqueous globular proteins Protein-solvent
Life Science Webinar Series
 Life Science Webinar Series Elegant protein- protein docking in Discovery Studio Francisco Hernandez-Guzman, Ph.D. November 20, 2007 Sr. Solutions Scientist fhernandez@accelrys.com Agenda In silico protein-protein
Life Science Webinar Series Elegant protein- protein docking in Discovery Studio Francisco Hernandez-Guzman, Ph.D. November 20, 2007 Sr. Solutions Scientist fhernandez@accelrys.com Agenda In silico protein-protein
Shortest Lattice Vector Enumeration on Graphics Cards
 Shortest Lattice Vector Enumeration on Graphics Cards Jens Hermans 1 Michael Schneider 2 Fréderik Vercauteren 1 Johannes Buchmann 2 Bart Preneel 1 1 K.U.Leuven 2 TU Darmstadt SHARCS - 10 September 2009
Shortest Lattice Vector Enumeration on Graphics Cards Jens Hermans 1 Michael Schneider 2 Fréderik Vercauteren 1 Johannes Buchmann 2 Bart Preneel 1 1 K.U.Leuven 2 TU Darmstadt SHARCS - 10 September 2009
Structure Investigation of Fam20C, a Golgi Casein Kinase
 Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Identifying Interaction Hot Spots with SuperStar
 Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Towards fast and accurate binding affinity. prediction with pmemdgti: an efficient. implementation of GPU-accelerated. Thermodynamic Integration
 Towards fast and accurate binding affinity prediction with pmemdgti: an efficient implementation of GPU-accelerated Thermodynamic Integration Tai-Sung Lee,, Yuan Hu, Brad Sherborne, Zhuyan Guo, and Darrin
Towards fast and accurate binding affinity prediction with pmemdgti: an efficient implementation of GPU-accelerated Thermodynamic Integration Tai-Sung Lee,, Yuan Hu, Brad Sherborne, Zhuyan Guo, and Darrin
ONETEP PB/SA: Application to G-Quadruplex DNA Stability. Danny Cole
 ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
Using AutoDock 4 with ADT: A Tutorial
 Using AutoDock 4 with ADT: A Tutorial Ruth Huey Sargis Dallakyan Alex Perryman David S. Goodsell (Garrett Morris) 9/2/08 Using AutoDock 4 with ADT 1 What is Docking? Predicting the best ways two molecules
Using AutoDock 4 with ADT: A Tutorial Ruth Huey Sargis Dallakyan Alex Perryman David S. Goodsell (Garrett Morris) 9/2/08 Using AutoDock 4 with ADT 1 What is Docking? Predicting the best ways two molecules
Adaptive Heterogeneous Computing with OpenCL: Harnessing hundreds of GPUs and CPUs
 Adaptive Heterogeneous Computing with OpenCL: Harnessing hundreds of GPUs and CPUs Simon McIntosh-Smith simonm@cs.bris.ac.uk Head of Microelectronics Research University of Bristol, UK 1 ! A brief biography
Adaptive Heterogeneous Computing with OpenCL: Harnessing hundreds of GPUs and CPUs Simon McIntosh-Smith simonm@cs.bris.ac.uk Head of Microelectronics Research University of Bristol, UK 1 ! A brief biography
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
D3RGC2: free energy scoring by alchemical free energy implementation in SOMD
 D3RGC2: free energy scoring by alchemical free energy implementation in SOMD Julien Michel EaStCHEM School of Chemistry, University of Edinburgh, United Kingdom D3R webinar 27/03/17 Acknowledgements: group
D3RGC2: free energy scoring by alchemical free energy implementation in SOMD Julien Michel EaStCHEM School of Chemistry, University of Edinburgh, United Kingdom D3R webinar 27/03/17 Acknowledgements: group
Molecular Dynamics Simulations
 MDGRAPE-3 chip: A 165- Gflops application-specific LSI for Molecular Dynamics Simulations Makoto Taiji High-Performance Biocomputing Research Team Genomic Sciences Center, RIKEN Molecular Dynamics Simulations
MDGRAPE-3 chip: A 165- Gflops application-specific LSI for Molecular Dynamics Simulations Makoto Taiji High-Performance Biocomputing Research Team Genomic Sciences Center, RIKEN Molecular Dynamics Simulations
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
interactions and reaction mechansims in peroxidases and aldo-keto reductases Carme Rovira ICREA / Parc Científic de Barcelona Barcelona
 Molecular modeling of enzyme-substrate interactions and reaction mechansims in peroxidases and aldo-keto reductases Carme Rovira ICREA / Parc Científic de Barcelona Barcelona What we do Molecular (atomistic)
Molecular modeling of enzyme-substrate interactions and reaction mechansims in peroxidases and aldo-keto reductases Carme Rovira ICREA / Parc Científic de Barcelona Barcelona What we do Molecular (atomistic)
Virtual affinity fingerprints in drug discovery: The Drug Profile Matching method
 Ágnes Peragovics Virtual affinity fingerprints in drug discovery: The Drug Profile Matching method PhD Theses Supervisor: András Málnási-Csizmadia DSc. Associate Professor Structural Biochemistry Doctoral
Ágnes Peragovics Virtual affinity fingerprints in drug discovery: The Drug Profile Matching method PhD Theses Supervisor: András Málnási-Csizmadia DSc. Associate Professor Structural Biochemistry Doctoral
Topology based deep learning for biomolecular data
 Topology based deep learning for biomolecular data Guowei Wei Departments of Mathematics Michigan State University http://www.math.msu.edu/~wei American Institute of Mathematics July 23-28, 2017 Grant
Topology based deep learning for biomolecular data Guowei Wei Departments of Mathematics Michigan State University http://www.math.msu.edu/~wei American Institute of Mathematics July 23-28, 2017 Grant
Computational Biochemistry in the New Era of HPC. Imran Haque Department of Computer Science Stanford University
 Folding@Everywhere Computational Biochemistry in the New Era of HPC Imran Haque Department of Computer Science Stanford University http://cs.stanford.edu/people/ihaque http://folding.stanford.edu Hyperience,
Folding@Everywhere Computational Biochemistry in the New Era of HPC Imran Haque Department of Computer Science Stanford University http://cs.stanford.edu/people/ihaque http://folding.stanford.edu Hyperience,
High Performance Computing
 High Performance Computing ADVANCED SCIENTIFIC COMPUTING Dr. Ing. Morris Riedel Adjunct Associated Professor School of Engineering and Natural Sciences, University of Iceland Research Group Leader, Juelich
High Performance Computing ADVANCED SCIENTIFIC COMPUTING Dr. Ing. Morris Riedel Adjunct Associated Professor School of Engineering and Natural Sciences, University of Iceland Research Group Leader, Juelich
Hybrid Vigor. Using Heterogeneous HPC to Accelerate Chemical Biology. Imran Haque Department of Computer Science Stanford University
 Hybrid Vigor Using Heterogeneous HPC to Accelerate Chemical Biology Imran Haque Department of Computer Science Stanford University http://cs.stanford.edu/people/ihaque http://folding.stanford.edu Bio-Molecular
Hybrid Vigor Using Heterogeneous HPC to Accelerate Chemical Biology Imran Haque Department of Computer Science Stanford University http://cs.stanford.edu/people/ihaque http://folding.stanford.edu Bio-Molecular
Parallel Asynchronous Hybrid Krylov Methods for Minimization of Energy Consumption. Langshi CHEN 1,2,3 Supervised by Serge PETITON 2
 1 / 23 Parallel Asynchronous Hybrid Krylov Methods for Minimization of Energy Consumption Langshi CHEN 1,2,3 Supervised by Serge PETITON 2 Maison de la Simulation Lille 1 University CNRS March 18, 2013
1 / 23 Parallel Asynchronous Hybrid Krylov Methods for Minimization of Energy Consumption Langshi CHEN 1,2,3 Supervised by Serge PETITON 2 Maison de la Simulation Lille 1 University CNRS March 18, 2013
Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation
 Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation Charles Blundell charles.blundell@c4xdiscovery.com www.c4xdiscovery.com Rigid: single
Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation Charles Blundell charles.blundell@c4xdiscovery.com www.c4xdiscovery.com Rigid: single