Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation
|
|
|
- Douglas Golden
- 6 years ago
- Views:
Transcription
1 Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation Charles Blundell
2 Rigid: single conformation Flexibility requires ensembles All data satisfied by a single conformation Flexible: two+ rapidly interchanging conformations Fitting data to single conformation produces unrealistic virtual conformations Replace MEAN averaging MODAL averaging Data needs to be fit to an ensemble of conformations as a better model of reality
3 mean angle Ensemble generation
4 Ensemble generation mean angle libration 0º 0º 270º 90º 270º 90º 180º large libration 180º small libration
5 Ensemble generation mean angle libration 3 rd minor mode 0º 0º 270º 90º 270º 90º 2 nd mode with its own mean & libration 180º large libration 180º small libration Single conformation model requires 1 variable per torsion C4XD dynamic model requires up to 8 variables per torsion (3 means, 3 librations, 2 population partitions) Avoids the problems of ensemble generation via comp chem Therefore ~10x more data is needed to solve the structure
6 Data and fitting 1) Combine multiple kinds of data into one coherent solution: NOESY/ROESY! Scalar couplings, Residual Dipolar Couplings Chemical shifts, H-bonds etc. etc. 2) Make the most of what you can measure:!! Explicitly calculate exact value of all observables! NOE x-peaks heights including nonoes (height = zero) Avoid approximations and assumptions inherent in e.g. converting NOES into distances, 3 JHH into torsional angles 3) Fitting function similar to that used in X-ray crystallography:!2 least-squares measure!! goodness of fit between measured data and theoretical prediction allowing for both measurement and prediction errors iterative rounds varying dynamic model until best fit found for fewest number of variables Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation Blundell CD, Packer MJ and Almond A, Bioorganic & Medicinal Chemistry, 21 (17),


7 Software implementation NMR data Proprietary software suite 4D-structure 2-3 weeks




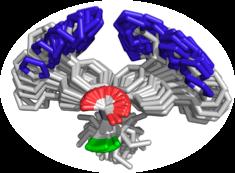
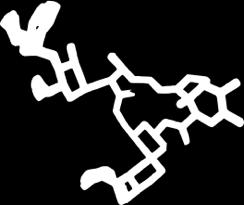

8 Applicable to all ligands Lisinopril Carazolol DRUGS Hyaluronan CARBOHYDRATES Losartan Ivermectin Streptomycin PEPTIDES Methotrexate AngiotensinII TRH COFACTORS Acetyl-CoA
9 Streptomycin solution structure Data type No. of restraints NOESY NOEs 176 no-noes 93 Residual dipolar couplings 4.5-6% gel 35 Scalar couplings TOTAL restraints per rotatable bond Composition of NOE restraints Classification NOEs nonoes All Intraresidue S R G TOTAL Data recorded in water at physiological temperature and ph Interresidue S1-R R2-G S1-G TOTAL
10 4D-structure Mean positions for each conformational mode Full ensemble showing range of libration adopted in solution
11 2 conformational families Glycosidic linkage between ribose and glucosamine adopts two distinct conformations * * MAJOR 62 ± 16 % MINOR 38 ± 16 %

12 Linkage geometry distribution "#$%!"&$%!'$% $% '$% "&$% "#$ "#$%!"&$%!'$% $% '$% "&$% "#$%
 10 structures")

13 Crystal structures Ribosome (natural target) 10 structures Transcriptional regulator TcaR 2 structures RNA aptamers 2 structures



14 Free vs primary bioactive "#$%!"&$%!'$% $% '$% "&$% "#$% "#$%!"&$%!'$% $% '$% "&$% "#$ Family 1 "#$%!"&$%!'$% $% '$% "&$% "#$%
15 Free vs other bioactives RNA aptamers 2 structures family 1 & family 2 Transcriptional regulator TcaR 2 structures family 1 & family 2 "#$%!"&$%!'$% $% '$% "&$% "#$ Family 2 Family 2 "#$%!"&$%!'$% $% '$% "&$% "#$%

16 Free vs other bioactives RNA aptamers 2 structures family 1 & family 2 Transcriptional regulator TcaR 2 structures family 1 & family 2 Di!erences between free and bioactive conformations impact observed binding a"nity Free solution structure predicts the diverse range of bioactive forms "#$%!"&$%!'$% $% '$% "&$% "#$ Family 2 Family 2 "#$%!"&$%!'$% $% '$% "&$% "#$%


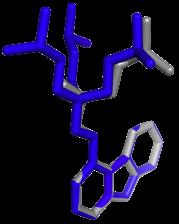
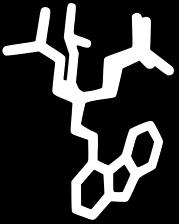

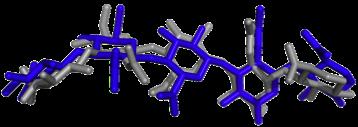
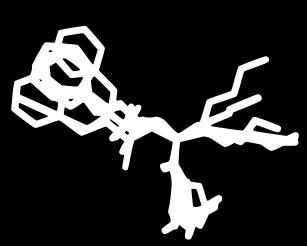

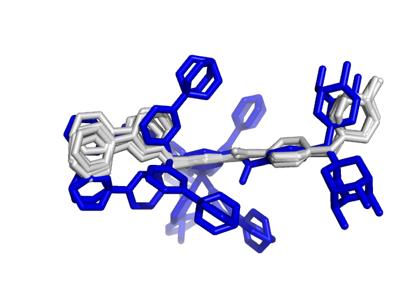
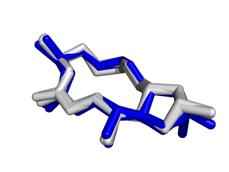
17 Modes vs Bioactive Conformation(s) BrefeldinA Carazolol Streptomycin Ivermectin Lisinopril Imatinib Hyaluronan

/$&/$1,34$1&'()*+,Jonathan Byrne*, Thorsten Nowak, Barrie Martin & Charles Blundell C4X")





.")










18 In-house drug discovery Currently running several programs HI, H2L and LO phases Type A & B GPCRs, ion channels, PPI With and without co-crystal support Free Ligand Conformational Populations in Solution A Powerful Drug Discovery Tool!"#$%&'()*+,-.$/+0$1&2+/'&)/$&/$1,34$1&'()*+,Jonathan Byrne*, Thorsten Nowak, Barrie Martin & Charles Blundell C4X Discovery Ltd., Unit 310 Ducie House, 37 Ducie Street, Manchester, M1 2JW, UK; Contact Jonathan.Byrne@C4XDiscovery.com The dynamic interchange of accessible low energy conformations for ligand molecules has traditionally been predicted computationally or extrapolated from small molecule crystal structure data. Here we present a new quantitative and precise NMR methodology that provides experimentally-derived detailed conformational information for free ligands in aqueous solution. These 4D-structures give unprecedented insight into how to exploit and control ligand conformational preferences in drug design. The detailed conformational information on ligand molecules contained in 4D-structures can be applied to all stages and settings of the drug discovery process. In Hit Identification, accurate 3D-pharmcophore information derived from reference compounds permits the rapid identification of chemical equity independently of other screening approaches (e.g., VS, HTS). In Lead-Generation and Lead-Optimisation, 4D-structures can significantly impact compound design separately or in conjunction with traditional structure-based drug design approaches. Specific examples for a range of targets are presented, demonstrating how 4D-structures can be used to great effect in drug discovery. Case study Orexin-1 selective compounds 9 Hit-to-Lead *+,-+./+0!'( /+0!'(1 4,05%!'() #$%&!'() 7 6 Target Area 5 " 6 67+!8-0$3-$0+8!9:!-758!,;<+%!5%%$8-0;-+!-7+!39<3+,-!;<=!=9!<9-!0+,0+8+<-!-7+! &!=+>+%9,+=!;-!?@A!25839>+0& Consensus Ph4 Shape-o-phore + Project Start, 6 4D structures -> VS 38 compounds tested, 5 < 1µM & de novo design! Lead Generation Problem Solving 7 8 pic50 Orexin 1 9 Potent and Selective Compounds Design exploiting Ph4 & Shape-o-phore 86 compounds, 1 Synth. Chem., 1 Med. Chem., 9 months Where in drug design do ligand 4D-structures add value? 15% 85% Libration Major Modes 4K5Y1 Consensus Ph4 Elements Conformational Dynamics Complementing SBDD Ph4 Positive impact on: Project initiation, SBDD, Comp. Chem., potency optimisation; Sel. Improvements, Med. Chem. problem solving Shape-o-phore defines the steric and electronic factors which determine the conformational dynamics & selection in solution which related to affinity Comparing and contrasting bound v free solution structure impacts positively on SBDD How do we measure ligand 4D-structures? bimodal population 50:50 unimodal angle: -77±8 libration: 25±7 trimodal 56:38:6 NOE, ROE RDC, 3J,!" Small Molecule NMR coupled with proprietary software generates 243 modes Occupancy Developed 3 novel series of <10 nm potent and 1000-fold orexin-1 selective antagonists 86 compounds 1 Synth. Chem. 1 Med. Chem. 9 months pic50 Orexin % in 1 of 27 modes 45% in 1 of 9 modes Conformation index an experimentally determined dynamic solution structure which is predictive of the bioactive conformation NMR-based 4D solution structures map the complete conformational space a small molecule naturally oscillates through in solution Dynamic solution structures are resolved into prioritised!idealised" conformers (shapes) 56 7)88+/'9+&/:$;<:$;+=/:$><:$?),9)8=9):$.<:$!@+/4:$A<$;<$B<:$%),C:$.<$D<:$>=E=-+,&:$.<:$F=,'@=88:$G<$7<$HIJ5K6:$!"#$%&:$'(()HL"MN6:$"KOP"K< I6?83/1+88:$!<$%<:$Q=(R+,:$F<$><:$S$.82)/1:$.<$HIJ5K6:$*+,,%-".+/)0)1&2+/+."3)/4&1+5#%6!"!78)H5L6:$"NOLP"NLT<$ C4X Discovery information All rights reserved. Worked examples see Poster 18
19 Using 4D structures in design In the absence of co-crystal data 4D-structures provide an estimate of the bioactive conformation Comparison with di"erent sca"olds reduces uncertainties Potency optimisation by tracking changes vs activity until Ph4 is known Synergistic with co-crystal data Di"erences between free and bioactive conformations directly address potency 0º 0º 270º 90º 270º 90º 180º adjust populations 180º adjust means Database of solution conformational behaviour ~1000 bonds, ~100 structures de novo conformational design and isostere replacement

20 Summary Accurate quantification of free ligand conformational preferences by NMR New ensemble description minimising number of new variables Increased amounts of experimental data Software supported : 2-3 weeks per structure Applicable to all classes of ligands Their relationship to the bioactive conformation 4D-structures frequently predict the bioactive conformation Can therefore be used to drive HI, H2L and LO processes Can be used independently of or synergistically with co-crystal data charles.blundell@c4xdiscovery.com
C4XD.L Conformetrics and its applica6on in drug discovery
 C4XD.L Conformetrics and its applica6on in drug discovery Dr Thorsten owak; 1 st Anglo-ordic-MedChem Conference 11 th 14 th June 2017 1 CTET Conformetrics, what it is and how its used in drug discovery
C4XD.L Conformetrics and its applica6on in drug discovery Dr Thorsten owak; 1 st Anglo-ordic-MedChem Conference 11 th 14 th June 2017 1 CTET Conformetrics, what it is and how its used in drug discovery
Toward an Understanding of GPCR-ligand Interactions. Alexander Heifetz
 Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
Hit Finding and Optimization Using BLAZE & FORGE
 Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
FRAGMENT SCREENING IN LEAD DISCOVERY BY WEAK AFFINITY CHROMATOGRAPHY (WAC )
") FRAGMENT SCREENING IN LEAD DISCOVERY BY WEAK AFFINITY CHROMATOGRAPHY (WAC ) SARomics Biostructures AB & Red Glead Discovery AB Medicon Village, Lund, Sweden Fragment-based lead discovery The basic idea:
FRAGMENT SCREENING IN LEAD DISCOVERY BY WEAK AFFINITY CHROMATOGRAPHY (WAC ) SARomics Biostructures AB & Red Glead Discovery AB Medicon Village, Lund, Sweden Fragment-based lead discovery The basic idea:
CSD. Unlock value from crystal structure information in the CSD
 CSD CSD-System Unlock value from crystal structure information in the CSD The Cambridge Structural Database (CSD) is the world s most comprehensive and up-todate knowledge base of crystal structure data,
CSD CSD-System Unlock value from crystal structure information in the CSD The Cambridge Structural Database (CSD) is the world s most comprehensive and up-todate knowledge base of crystal structure data,
In Silico Investigation of Off-Target Effects
 PHARMA & LIFE SCIENCES WHITEPAPER In Silico Investigation of Off-Target Effects STREAMLINING IN SILICO PROFILING In silico techniques require exhaustive data and sophisticated, well-structured informatics
PHARMA & LIFE SCIENCES WHITEPAPER In Silico Investigation of Off-Target Effects STREAMLINING IN SILICO PROFILING In silico techniques require exhaustive data and sophisticated, well-structured informatics
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Experimental Techniques in Protein Structure Determination
 Experimental Techniques in Protein Structure Determination Homayoun Valafar Department of Computer Science and Engineering, USC Two Main Experimental Methods X-Ray crystallography Nuclear Magnetic Resonance
Experimental Techniques in Protein Structure Determination Homayoun Valafar Department of Computer Science and Engineering, USC Two Main Experimental Methods X-Ray crystallography Nuclear Magnetic Resonance
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Structure-based maximal affinity model predicts small-molecule druggability
 Structure-based maximal affinity model predicts small-molecule druggability Alan Cheng alan.cheng@amgen.com IMA Workshop (Jan 17, 2008) Druggability prediction Introduction Affinity model Some results
Structure-based maximal affinity model predicts small-molecule druggability Alan Cheng alan.cheng@amgen.com IMA Workshop (Jan 17, 2008) Druggability prediction Introduction Affinity model Some results
Implementation of novel tools to facilitate fragment-based drug discovery by NMR:
 Implementation of novel tools to facilitate fragment-based drug discovery by NMR: Automated analysis of large sets of ligand-observed NMR binding data and 19 F methods Andreas Lingel Global Discovery Chemistry
Implementation of novel tools to facilitate fragment-based drug discovery by NMR: Automated analysis of large sets of ligand-observed NMR binding data and 19 F methods Andreas Lingel Global Discovery Chemistry
Structure-Based Drug Discovery An Overview
 Structure-Based Drug Discovery An Overview Edited by Roderick E. Hubbard University of York, Heslington, York, UK and Vernalis (R&D) Ltd, Abington, Cambridge, UK RSC Publishing Contents Chapter 1 3D Structure
Structure-Based Drug Discovery An Overview Edited by Roderick E. Hubbard University of York, Heslington, York, UK and Vernalis (R&D) Ltd, Abington, Cambridge, UK RSC Publishing Contents Chapter 1 3D Structure
Protein Structure Determination using NMR Spectroscopy. Cesar Trinidad
 Protein Structure Determination using NMR Spectroscopy Cesar Trinidad Introduction Protein NMR Involves the analysis and calculation of data collected from multiple NMR techniques Utilizes Nuclear Magnetic
Protein Structure Determination using NMR Spectroscopy Cesar Trinidad Introduction Protein NMR Involves the analysis and calculation of data collected from multiple NMR techniques Utilizes Nuclear Magnetic
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Interpreting and evaluating biological NMR in the literature. Worksheet 1
 Interpreting and evaluating biological NMR in the literature Worksheet 1 1D NMR spectra Application of RF pulses of specified lengths and frequencies can make certain nuclei detectable We can selectively
Interpreting and evaluating biological NMR in the literature Worksheet 1 1D NMR spectra Application of RF pulses of specified lengths and frequencies can make certain nuclei detectable We can selectively
Ping-Chiang Lyu. Institute of Bioinformatics and Structural Biology, Department of Life Science, National Tsing Hua University.
 Pharmacophore-based Drug design Ping-Chiang Lyu Institute of Bioinformatics and Structural Biology, Department of Life Science, National Tsing Hua University 96/08/07 Outline Part I: Analysis The analytical
Pharmacophore-based Drug design Ping-Chiang Lyu Institute of Bioinformatics and Structural Biology, Department of Life Science, National Tsing Hua University 96/08/07 Outline Part I: Analysis The analytical
Protein Structure Prediction and Protein-Ligand Docking
 Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Proteins are not rigid structures: Protein dynamics, conformational variability, and thermodynamic stability
 Proteins are not rigid structures: Protein dynamics, conformational variability, and thermodynamic stability Dr. Andrew Lee UNC School of Pharmacy (Div. Chemical Biology and Medicinal Chemistry) UNC Med
Proteins are not rigid structures: Protein dynamics, conformational variability, and thermodynamic stability Dr. Andrew Lee UNC School of Pharmacy (Div. Chemical Biology and Medicinal Chemistry) UNC Med
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Data Quality Issues That Can Impact Drug Discovery
 Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Notes of Dr. Anil Mishra at 1
 Introduction Quantitative Structure-Activity Relationships QSPR Quantitative Structure-Property Relationships What is? is a mathematical relationship between a biological activity of a molecular system
Introduction Quantitative Structure-Activity Relationships QSPR Quantitative Structure-Property Relationships What is? is a mathematical relationship between a biological activity of a molecular system
Quantum Mechanical Models of P450 Metabolism to Guide Optimization of Metabolic Stability
 Quantum Mechanical Models of P450 Metabolism to Guide Optimization of Metabolic Stability Optibrium Webinar 2015, June 17 2015 Jonathan Tyzack, Matthew Segall, Peter Hunt Optibrium, StarDrop, Auto-Modeller
Quantum Mechanical Models of P450 Metabolism to Guide Optimization of Metabolic Stability Optibrium Webinar 2015, June 17 2015 Jonathan Tyzack, Matthew Segall, Peter Hunt Optibrium, StarDrop, Auto-Modeller
QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression
 APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor
APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor
Virtual Screening: How Are We Doing?
 Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
The use of Design of Experiments to develop Efficient Arrays for SAR and Property Exploration
 The use of Design of Experiments to develop Efficient Arrays for SAR and Property Exploration Chris Luscombe, Computational Chemistry GlaxoSmithKline Summary of Talk Traditional approaches SAR Free-Wilson
The use of Design of Experiments to develop Efficient Arrays for SAR and Property Exploration Chris Luscombe, Computational Chemistry GlaxoSmithKline Summary of Talk Traditional approaches SAR Free-Wilson
Structure based drug design and LIE models for GPCRs
 Structure based drug design and LIE models for GPCRs Peter Kolb kolb@docking.org Shoichet Lab ACS 237 th National Meeting, March 24, 2009 p.1/26 [Acknowledgements] Brian Shoichet John Irwin Mike Keiser
Structure based drug design and LIE models for GPCRs Peter Kolb kolb@docking.org Shoichet Lab ACS 237 th National Meeting, March 24, 2009 p.1/26 [Acknowledgements] Brian Shoichet John Irwin Mike Keiser
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
DNA Structure. Voet & Voet: Chapter 29 Pages Slide 1
 DNA Structure Voet & Voet: Chapter 29 Pages 1107-1122 Slide 1 Review The four DNA bases and their atom names The four common -D-ribose conformations All B-DNA ribose adopt the C2' endo conformation All
DNA Structure Voet & Voet: Chapter 29 Pages 1107-1122 Slide 1 Review The four DNA bases and their atom names The four common -D-ribose conformations All B-DNA ribose adopt the C2' endo conformation All
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors
 MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery
 21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
The Chemistry department approved by the American Chemical Society offers a Chemistry degree in the following concentrations:
 Chemistry 1 Chemistry 203-C Materials Science Building Telephone: 256.824.6153 Email: chem.admin@uah.edu (chem@uah.edu) The Chemistry department approved by the American Chemical Society offers a Chemistry
Chemistry 1 Chemistry 203-C Materials Science Building Telephone: 256.824.6153 Email: chem.admin@uah.edu (chem@uah.edu) The Chemistry department approved by the American Chemical Society offers a Chemistry
CSD. CSD-Enterprise. Access the CSD and ALL CCDC application software
 CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
NMR in Medicine and Biology
 NMR in Medicine and Biology http://en.wikipedia.org/wiki/nmr_spectroscopy MRI- Magnetic Resonance Imaging (water) In-vivo spectroscopy (metabolites) Solid-state t NMR (large structures) t Solution NMR
NMR in Medicine and Biology http://en.wikipedia.org/wiki/nmr_spectroscopy MRI- Magnetic Resonance Imaging (water) In-vivo spectroscopy (metabolites) Solid-state t NMR (large structures) t Solution NMR
Important Aspects of Fragment Screening Collection Design
 Important Aspects of Fragment Screening Collection Design Phil Cox, Ph. D., Discovery Chemistry and Technology, AbbVie, USA Cresset User Group Meeting, Cambridge UK. Thursday, June 29 th 2017 Disclosure-
Important Aspects of Fragment Screening Collection Design Phil Cox, Ph. D., Discovery Chemistry and Technology, AbbVie, USA Cresset User Group Meeting, Cambridge UK. Thursday, June 29 th 2017 Disclosure-
Targeting protein-protein interactions: A hot topic in drug discovery
 Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
NMR, X-ray Diffraction, Protein Structure, and RasMol
 NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design
 Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Everyday NMR. Innovation with Integrity. Why infer when you can be sure? NMR
 Everyday NMR Why infer when you can be sure? Innovation with Integrity NMR Only NMR gives you definitive answers, on your terms. Over the past half-century, scientists have used nuclear magnetic resonance
Everyday NMR Why infer when you can be sure? Innovation with Integrity NMR Only NMR gives you definitive answers, on your terms. Over the past half-century, scientists have used nuclear magnetic resonance
Theory and Applications of Residual Dipolar Couplings in Biomolecular NMR
 Theory and Applications of Residual Dipolar Couplings in Biomolecular NMR Residual Dipolar Couplings (RDC s) Relatively new technique ~ 1996 Nico Tjandra, Ad Bax- NIH, Jim Prestegard, UGA Combination of
Theory and Applications of Residual Dipolar Couplings in Biomolecular NMR Residual Dipolar Couplings (RDC s) Relatively new technique ~ 1996 Nico Tjandra, Ad Bax- NIH, Jim Prestegard, UGA Combination of
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Practical QSAR and Library Design: Advanced tools for research teams
 DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
Minireview: Molecular Structure and Dynamics of Drug Targets
 Prague Medical Report / Vol. 109 (2008) No. 2 3, p. 107 112 107) Minireview: Molecular Structure and Dynamics of Drug Targets Dahl S. G., Sylte I. Department of Pharmacology, Institute of Medical Biology,
Prague Medical Report / Vol. 109 (2008) No. 2 3, p. 107 112 107) Minireview: Molecular Structure and Dynamics of Drug Targets Dahl S. G., Sylte I. Department of Pharmacology, Institute of Medical Biology,
COMBINATORIAL CHEMISTRY: CURRENT APPROACH
 COMBINATORIAL CHEMISTRY: CURRENT APPROACH Dwivedi A. 1, Sitoke A. 2, Joshi V. 3, Akhtar A.K. 4* and Chaturvedi M. 1, NRI Institute of Pharmaceutical Sciences, Bhopal, M.P.-India 2, SRM College of Pharmacy,
COMBINATORIAL CHEMISTRY: CURRENT APPROACH Dwivedi A. 1, Sitoke A. 2, Joshi V. 3, Akhtar A.K. 4* and Chaturvedi M. 1, NRI Institute of Pharmaceutical Sciences, Bhopal, M.P.-India 2, SRM College of Pharmacy,
Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Applying Bioisosteric Transformations to Predict Novel, High Quality Compounds
 Applying Bioisosteric Transformations to Predict Novel, High Quality Compounds Dr James Chisholm,* Dr John Barnard, Dr Julian Hayward, Dr Matthew Segall*, Mr Edmund Champness*, Dr Chris Leeding,* Mr Hector
Applying Bioisosteric Transformations to Predict Novel, High Quality Compounds Dr James Chisholm,* Dr John Barnard, Dr Julian Hayward, Dr Matthew Segall*, Mr Edmund Champness*, Dr Chris Leeding,* Mr Hector
Statistical concepts in QSAR.
 Statistical concepts in QSAR. Computational chemistry represents molecular structures as a numerical models and simulates their behavior with the equations of quantum and classical physics. Available programs
Statistical concepts in QSAR. Computational chemistry represents molecular structures as a numerical models and simulates their behavior with the equations of quantum and classical physics. Available programs
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Hydrogen Bonding & Molecular Design Peter
 Hydrogen Bonding & Molecular Design Peter Kenny(pwk.pub.2008@gmail.com) Hydrogen Bonding in Drug Discovery & Development Interactions between drug and water molecules (Solubility, distribution, permeability,
Hydrogen Bonding & Molecular Design Peter Kenny(pwk.pub.2008@gmail.com) Hydrogen Bonding in Drug Discovery & Development Interactions between drug and water molecules (Solubility, distribution, permeability,
The Conformation Search Problem
 Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
Different conformations of the drugs within the virtual library of FDA approved drugs will be generated.
 Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
Molecular Modeling lecture 2
 Molecular Modeling 2018 -- lecture 2 Topics 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction 4. Where do protein structures come from? X-ray crystallography
Molecular Modeling 2018 -- lecture 2 Topics 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction 4. Where do protein structures come from? X-ray crystallography
BioSolveIT. A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities Ingo Dramburg BioSolve IT Gmb An der Ziegelei 75 53757
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities Ingo Dramburg BioSolve IT Gmb An der Ziegelei 75 53757
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Introduction solution NMR
 2 NMR journey Introduction solution NMR Alexandre Bonvin Bijvoet Center for Biomolecular Research with thanks to Dr. Klaartje Houben EMBO Global Exchange course, IHEP, Beijing April 28 - May 5, 20 3 Topics
2 NMR journey Introduction solution NMR Alexandre Bonvin Bijvoet Center for Biomolecular Research with thanks to Dr. Klaartje Houben EMBO Global Exchange course, IHEP, Beijing April 28 - May 5, 20 3 Topics
I690/B680 Structural Bioinformatics Spring Protein Structure Determination by NMR Spectroscopy
 I690/B680 Structural Bioinformatics Spring 2006 Protein Structure Determination by NMR Spectroscopy Suggested Reading (1) Van Holde, Johnson, Ho. Principles of Physical Biochemistry, 2 nd Ed., Prentice
I690/B680 Structural Bioinformatics Spring 2006 Protein Structure Determination by NMR Spectroscopy Suggested Reading (1) Van Holde, Johnson, Ho. Principles of Physical Biochemistry, 2 nd Ed., Prentice
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Building innovative drug discovery alliances
 Building innovative drug discovery alliances Hit optimisation o using fragments Mark kwhittaker Evotec AG, Fragments 2015, March 2015 Agenda Fragment optimisation in an ideal world Fragment optimisation
Building innovative drug discovery alliances Hit optimisation o using fragments Mark kwhittaker Evotec AG, Fragments 2015, March 2015 Agenda Fragment optimisation in an ideal world Fragment optimisation
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today
 est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
BioSolveIT. A Combinatorial Approach for Handling of Protonation and Tautomer Ambiguities in Docking Experiments
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
Building innovative drug discovery alliances. ovel histamine GPCR family antagonists by ragment screening and molecular modelling
 Building innovative drug discovery alliances ovel histamine GPCR family antagonists by ragment screening and molecular modelling votec AG, GPCRs; Fragments & Modelling, Sept. 20th, 2010 Outline of Presentation
Building innovative drug discovery alliances ovel histamine GPCR family antagonists by ragment screening and molecular modelling votec AG, GPCRs; Fragments & Modelling, Sept. 20th, 2010 Outline of Presentation
Introduction to" Protein Structure
 Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Use of data mining and chemoinformatics in the identification and optimization of high-throughput screening hits for NTDs
 Use of data mining and chemoinformatics in the identification and optimization of high-throughput screening hits for NTDs James Mills; Karl Gibson, Gavin Whitlock, Paul Glossop, Jean-Robert Ioset, Leela
Use of data mining and chemoinformatics in the identification and optimization of high-throughput screening hits for NTDs James Mills; Karl Gibson, Gavin Whitlock, Paul Glossop, Jean-Robert Ioset, Leela
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining
![Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining](/thumbs/91/107031261.jpg "Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining") Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
Copyright Mark Brandt, Ph.D A third method, cryogenic electron microscopy has seen increasing use over the past few years.
 Structure Determination and Sequence Analysis The vast majority of the experimentally determined three-dimensional protein structures have been solved by one of two methods: X-ray diffraction and Nuclear
Structure Determination and Sequence Analysis The vast majority of the experimentally determined three-dimensional protein structures have been solved by one of two methods: X-ray diffraction and Nuclear
1. 3-hour Open book exam. No discussion among yourselves.
 Lecture 13 Review 1. 3-hour Open book exam. No discussion among yourselves. 2. Simple calculations. 3. Terminologies. 4. Decriptive questions. 5. Analyze a pulse program using density matrix approach (omonuclear
Lecture 13 Review 1. 3-hour Open book exam. No discussion among yourselves. 2. Simple calculations. 3. Terminologies. 4. Decriptive questions. 5. Analyze a pulse program using density matrix approach (omonuclear
GC and CELPP: Workflows and Insights
 GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
Introduction to FBDD Fragment screening methods and library design
 Introduction to FBDD Fragment screening methods and library design Samantha Hughes, PhD Fragments 2013 RSC BMCS Workshop 3 rd March 2013 Copyright 2013 Galapagos NV Why fragment screening methods? Guess
Introduction to FBDD Fragment screening methods and library design Samantha Hughes, PhD Fragments 2013 RSC BMCS Workshop 3 rd March 2013 Copyright 2013 Galapagos NV Why fragment screening methods? Guess
Full wwpdb NMR Structure Validation Report i
 Full wwpdb NMR Structure Validation Report i Feb 17, 2018 06:22 am GMT PDB ID : 141D Title : SOLUTION STRUCTURE OF A CONSERVED DNA SEQUENCE FROM THE HIV-1 GENOME: RESTRAINED MOLECULAR DYNAMICS SIMU- LATION
Full wwpdb NMR Structure Validation Report i Feb 17, 2018 06:22 am GMT PDB ID : 141D Title : SOLUTION STRUCTURE OF A CONSERVED DNA SEQUENCE FROM THE HIV-1 GENOME: RESTRAINED MOLECULAR DYNAMICS SIMU- LATION
FRAUNHOFER IME SCREENINGPORT
 FRAUNHOFER IME SCREENINGPORT Design of screening projects General remarks Introduction Screening is done to identify new chemical substances against molecular mechanisms of a disease It is a question of
FRAUNHOFER IME SCREENINGPORT Design of screening projects General remarks Introduction Screening is done to identify new chemical substances against molecular mechanisms of a disease It is a question of
Analyzing Molecular Conformations Using the Cambridge Structural Database. Jason Cole Cambridge Crystallographic Data Centre
 Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses
 Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
Lecture for Molekylär bioinformatik X3 Feb Computational Chemistry in Drug Discovery. Mats Kihlén. Head of Research Informatics.
 Lecture for Molekylär bioinformatik X3 Feb 24 2004 Computational Chemistry in Drug Discovery Mats Kihlén Head of Research Informatics Biovitrum AB verview» The role of computational chemistry» Basic concepts
Lecture for Molekylär bioinformatik X3 Feb 24 2004 Computational Chemistry in Drug Discovery Mats Kihlén Head of Research Informatics Biovitrum AB verview» The role of computational chemistry» Basic concepts
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Early Stages of Drug Discovery in the Pharmaceutical Industry
 Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
CHEMISTRY (CHEM) CHEM 5800 Principles Of Materials Chemistry. Tutorial in selected topics in materials chemistry. S/U grading only.
 CHEM 5800 Principles Of Materials Chemistry. Tutorial in selected topics in materials chemistry. S/U grading only.") Chemistry (CHEM) 1 CHEMISTRY (CHEM) CHEM 5100 Principles of Organic and Inorganic Chemistry Study of coordination compounds with a focus on ligand bonding, electron counting, molecular orbital theory,
Chemistry (CHEM) 1 CHEMISTRY (CHEM) CHEM 5100 Principles of Organic and Inorganic Chemistry Study of coordination compounds with a focus on ligand bonding, electron counting, molecular orbital theory,
Molecular Modelling. Computational Chemistry Demystified. RSC Publishing. Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK
 Molecular Modelling Computational Chemistry Demystified Peter Bladon Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK John E. Gorton Gorton Systems, Glasgow, UK Robert B. Hammond Institute
Molecular Modelling Computational Chemistry Demystified Peter Bladon Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK John E. Gorton Gorton Systems, Glasgow, UK Robert B. Hammond Institute
Schrodinger ebootcamp #3, Summer EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist
 Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
MSc Drug Design. Module Structure: (15 credits each) Lectures and Tutorials Assessment: 50% coursework, 50% unseen examination.
 Lectures and Tutorials Assessment: 50% coursework, 50% unseen examination.") Module Structure: (15 credits each) Lectures and Assessment: 50% coursework, 50% unseen examination. Module Title Module 1: Bioinformatics and structural biology as applied to drug design MEDC0075 In the
Module Structure: (15 credits each) Lectures and Assessment: 50% coursework, 50% unseen examination. Module Title Module 1: Bioinformatics and structural biology as applied to drug design MEDC0075 In the
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit
 October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?!
 for structure refinement from X-ray diffraction data?!") 1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?! Visual analysis: Identification of ligand density from
1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?! Visual analysis: Identification of ligand density from
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
A primer on pharmacology pharmacodynamics
 A primer on pharmacology pharmacodynamics Drug binding & effect Universidade do Algarve Faro 2017 by Ferdi Engels, Ph.D. 1 Pharmacodynamics Relation with pharmacokinetics? dosage plasma concentration site
A primer on pharmacology pharmacodynamics Drug binding & effect Universidade do Algarve Faro 2017 by Ferdi Engels, Ph.D. 1 Pharmacodynamics Relation with pharmacokinetics? dosage plasma concentration site
COMBINATORIAL CHEMISTRY IN A HISTORICAL PERSPECTIVE
 NUE FEATURE T R A N S F O R M I N G C H A L L E N G E S I N T O M E D I C I N E Nuevolution Feature no. 1 October 2015 Technical Information COMBINATORIAL CHEMISTRY IN A HISTORICAL PERSPECTIVE A PROMISING
NUE FEATURE T R A N S F O R M I N G C H A L L E N G E S I N T O M E D I C I N E Nuevolution Feature no. 1 October 2015 Technical Information COMBINATORIAL CHEMISTRY IN A HISTORICAL PERSPECTIVE A PROMISING
Protein Structure Analysis and Verification. Course S Basics for Biosystems of the Cell exercise work. Maija Nevala, BIO, 67485U 16.1.
 Protein Structure Analysis and Verification Course S-114.2500 Basics for Biosystems of the Cell exercise work Maija Nevala, BIO, 67485U 16.1.2008 1. Preface When faced with an unknown protein, scientists
Protein Structure Analysis and Verification Course S-114.2500 Basics for Biosystems of the Cell exercise work Maija Nevala, BIO, 67485U 16.1.2008 1. Preface When faced with an unknown protein, scientists
Advanced Medicinal Chemistry SLIDES B
 Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm