CDK-Taverna: an open workflow environment for cheminformatics
|
|
|
- Arabella Moore
- 6 years ago
- Views:
Transcription
 Egon L Willighagen (egon.")
1 BMC Bioinformatics This Provisional PDF corresponds to the article as it appeared upon acceptance. Fully formatted PDF and full text (HTML) versions will be made available soon. CDK-Taverna: an open workflow environment for cheminformatics BMC Bioinformatics 2010, 11:159 doi: / Thomas Kuhn Egon L Willighagen (egon.willighagen@farmbio.uu.se) Achim Zielesny (achim.zielesny@fh-gelsenkirchen.de) Christoph Steinbeck (steinbeck@ebi.ac.uk) ISSN Article type Software Submission date 7 September 2009 Acceptance date 29 March 2010 Publication date 29 March 2010 Article URL Like all articles in BMC journals, this peer-reviewed article was published immediately upon acceptance. It can be downloaded, printed and distributed freely for any purposes (see copyright notice below). Articles in BMC journals are listed in PubMed and archived at PubMed Central. For information about publishing your research in BMC journals or any BioMed Central journal, go to Kuhn et al., licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License ( which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
2 CDK-Taverna: an open workflow environment for cheminformatics Thomas Kuhn 1, Egon L. Willighagen 2, Achim Zielesny 1 and Christoph Steinbeck 3 1 Institute for Bioinformatics and Cheminformatics, University of Applied Sciences of Gelsenkirchen, Recklinghausen, Germany 2 Department of Pharmaceutical Biosciences, Uppsala University, Uppsala, Sweden 3 Chemoinformatics and Metabolism, European Bioinformatics Institute (EBI), Cambridge, UK Thomas Kuhn - thomas.kuhn@fh-gelsenkirchen.de; Egon L. Willighagen - egon.willighagen@farmbio.uu.se; Achim Zielesny - achim.zielesny@fh-gelsenkirchen.de; Christoph Steinbeck - steinbeck@ebi.ac.uk; Corresponding author Abstract Background: Small molecules are of increasing interest for bioinformatics in areas such as metabolomics and drug discovery. The recent release of large open access chemistry databases generates a demand for flexible tools to process them and discover new knowledge. To freely support open science based on these data resources, it is desirable for the processing tools to be open source and available for everyone. Results: Here we describe a novel combination of the workflow engine Taverna and the cheminformatics library Chemistry Development Kit (CDK) resulting in a open source workflow solution for cheminformatics. We have implemented more than 160 different workers to handle specific cheminformatics tasks. We describe the applications of CDK-Taverna in various usage scenarios. Conclusions: The combination of the workflow engine Taverna and the Chemistry Development Kit provides the first open source cheminformatics workflow solution for the biosciences. With the Taverna-community working towards a more powerful workflow engine and a more user-friendly user interface, CDK-Taverna has the potential to become a free alternative to existing proprietary workflow tools. 1
3 Background Small molecules are of increasing interest for bioinformatics in areas such as metabolomics and drug discovery. The recent release of large open chemistry databases into the public domain [1 4] calls for flexible, open toolkits to process them. These databases and tools will, for the first time, create opportunities for academia and third-world countries to perform state-of-the-art open drug discovery and translational research endeavors so far a domain of the pharmaceutical industry. Commonly used in this context are workflow engines for cheminformatics, where numerous recurring tasks can be automated, including tasks for chemical data filtering, transformation, curation and migration workflows chemical documentation and information retrieval related workflows (structures, reactions, pharmacophores, object relational data etc.) data analysis workflows (statistics and clustering/machine learning for QSAR, diversity analysis etc.) The workflow paradigm allows scientists to flexibly create generic workflows using different kinds of data sources, filters and algorithms, which can later be adapted to changing needs. In order to achieve this, library methods are encapsulated in Lego TM -like building blocks which can be manipulated with a mouse or any pointing device in a graphical environment, relieving the scientist from the need to learn a programming language. Building blocks are connected by data pipelines to enable data flow between them, which is why pipelining is often used interchangeably for workflow. Workflows are increasingly used in cheminformatics research [5, 6]. Existing proprietary or semi-proprietary implementations of the workflow or pipeline paradigm in molecular informatics include Pipeline Pilot [7] from SciTegic, a subsidiary of Accelrys or the InforSense platform from InforSense [8]. Both are commercially well established but closed source products with a large variety of different functionality. KNIME [9] is a modular data exploration platform which uses a dual licensing model with the Aladdin free public license. It is developed by the group of Michael Berthold at the University of Konstanz, Germany. KNIME is based on the open source Eclipse platform. An overview of workflow systems in life sciences was recently given by Tiwari et al. [10]. In 2005 we started to integrate our open source cheminformatics library, the Chemistry Development Kit (CDK) [11, 12] with Taverna [13 15], a workflow environment with an extensible architecture, to produce CDK-Taverna, the first completely free [16] workflow solution for cheminformatics, which we present here. 2
4 It makes additional use of other open source components such as Bioclipse [17] for visualization of workflow results, and Pgchem::tigress [18] as an interface to the database back-end for storage of large data sets. Implementation The here introduced CDK-Taverna plugin takes advantage of the plug-in detection manager of Taverna for its installation. This manager requires a plug-in description XML file containing a plug-in name, a version number, a target Taverna version number, a repository location and a Maven-like Java package description, all provided by the plug-in s installation website: After adding this URL, the manager presents all available plug-in versions graphically to the user. In order to install the CDK-Taverna plug-in the user selects the desired version after which all necessary Java libraries are installed on-the-fly from the given installation website. The CDK-Taverna plug-in is written in Java is published under the GNU Lesser General Public License (LGPL). Version uses CDK revision Like Taverna itself the CDK-Taverna plug-in uses Maven 2 [19] as a build system. To integrate the CDK functionality, the plug-in makes use of the extension points provided by Taverna allowing dynamic discovery of the provided functionality. The following sections describe what extension points are used, and how molecular data is represented when flowing through the workflow. Taverna s extension points Taverna allows the execution of workflows linking together heterogeneous open services, applications or databases (remote or local, private or public, third-party or home-grown) [20]. For the integration of these different resource types Taverna provides various interfaces and protocols for its extension. For example, it allows for easy access to webservices through WSDL [21] and SOAP [22]. The CDK-Taverna plug-in, on the other hand, uses Taverna s local extension mechanism. For local extensions, Taverna provides a list of different Service Provider Interfaces (SPI), as given in Table 1. CDK-Taverna implements several of these, integrating CDK functionality as so-called Local Workers which run on the same machine as the Taverna installation. Full JavaDoc documentation of the plug-in s source code is available at All workers in CDK-Taverna implement the CDKLocalWorker interface. It is used for the detection of workers by the CDKScavenger class which itself implements the Taverna SPI org.embl.ebi.escience.scuflui.workbench.scavenger interface. Adding user interfaces for some of 3
5 the workers requires an extension of the AbstractCDKProcessorAction which again implements the Taverna SPI org.embl.ebi.escience.scuflui.spi.processoractionspi. The use of this SPI allows the addition of, for example, file chooser dialogs for workers like file reader or writer. The anatomy of a CDK-Taverna worker To create a CDK-Taverna worker the Java class of this worker has to implement the CDKLocalWorker Interface. This interface defines that every worker has to define the following methods: public Map <String, DataThing> execute(map String, DataThing inputmap) throws TaskExecutionException; public String[] inputnames(); public String[] inputtypes(); public String[] outputnames(); public String[] outputtypes(); The method inputnames and outputnames return the names of the ports of each worker whereas the inputtypes and outputtypes methods return the names for the Java object types with its package declaration e.g. java/java.util.list for a List. Within CDK-Taverna chemical structures are passed around using the Java object java/org.openscience.cdk.applications.taverna.cmlchemfile Results The CDK-Taverna plug-in currently provides 164 different cheminformatics workers. The fields of application of these workers are described in Table 2. These include workers for input and output (I/O) of various chemical files and line notations formats, databases, and descriptors for atoms, bonds and molecules. The miscellaneous workers are e.g. a substructure filter, an aromaticity detector, an atom typer or a reaction enumerator. Some of the workers are outlined as part of example workflows described below (for a complete list see [23] or In the following, we outline the application of CDK-Taverna with selected workflow scenarios. A larger list of workflows is available from MyExperiment.org [24]. Iteration over large data sets Cheminformatics by definition deals with the discovery of chemical knowledge from large data collections. Because these data sources are usually too large to be loaded into memory as a whole, it is needed to loop 4
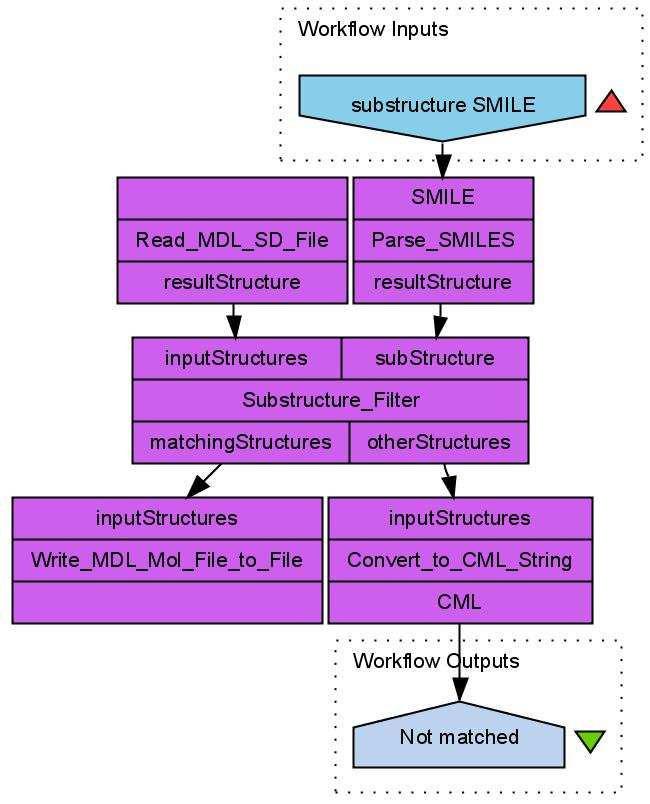
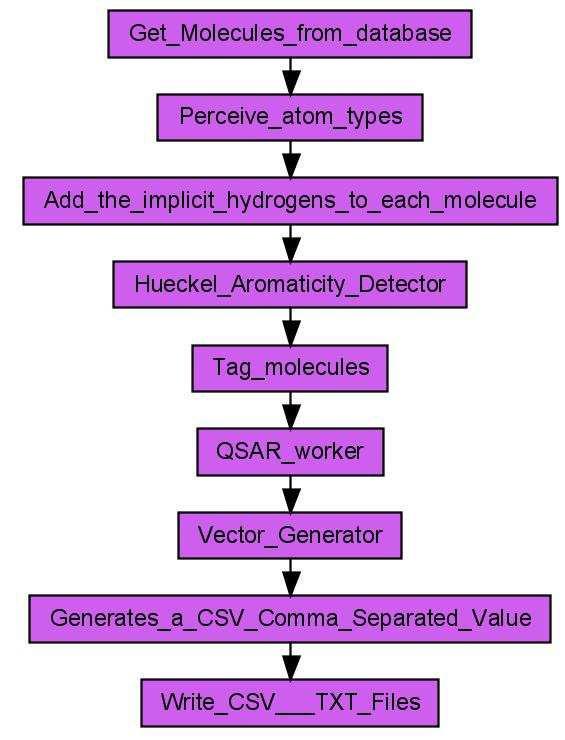
6 over all data entries to process them one by one. Unfortunately, the architecture of Taverna 1.7 does not support such loops. CDK-Taverna, therefore, provides workers which act like FOR or WHILE loops, making use of Taverna s iteration-and-retry mechanism to allow workflows to process large data sets. Database I/O For database support the CDK-Taverna project uses the PostgreSQL [25] relational database management system (RDBMS) with the open source Pgchem::tigress [18] extension. This combination allows storage and fast retrieval of up to a million molecules without running into memory limitations. The Pgchem::tigress extension uses an implementation of the Generalized Search Tree (GiST) [26] of the PostgreSQL database. CDK-Taverna can use a local installation of the PostgreSQL Database with the Pgchem::tigress extension or can connect to a remote instance. Scenario 1: Substructure Search We may want to design workflows for performing substructure searches in different ways depending on the type of input. In a first example the substructure workflow performs a topological substructure search on a list of given molecules and a given molecular substructure (see Figure 1). The workflow inputs are a molecular substructure represented in the SMILES line notation [27] and a list of structures stored in a MDL SDfile [28]. The structures which match the substructure are stored as MDL Molfiles [28]. The non-matching structures are converted into the Chemical Markup Language (CML) file format [29 31]. This small example workflow already combines four different molecular structure representations and the use of a topological substructure filter. A related workflow performs a substructure search directly on a database: It uses functionality provided by the Pgchem extension of the PostgreSQL database. This extension allows the use of SQL commands to perform a substructure search. A demonstration is given with the workflow in Figure 2. The molecules containing the substructure are reported in tabular form in a PDF file. Scenario 2: Descriptor Calculation The descriptor calculation workflow, depicted in Figure 3, starts with loading its molecules from a PostgreSQL database. The recognition of the atom types is the next step, followed by the addition of implicit hydrogens for each molecule as well as the detection of Hückel aromaticity. Each molecule is tagged for the descriptor calculation process. The tagging of the molecules is used to add an universal 5
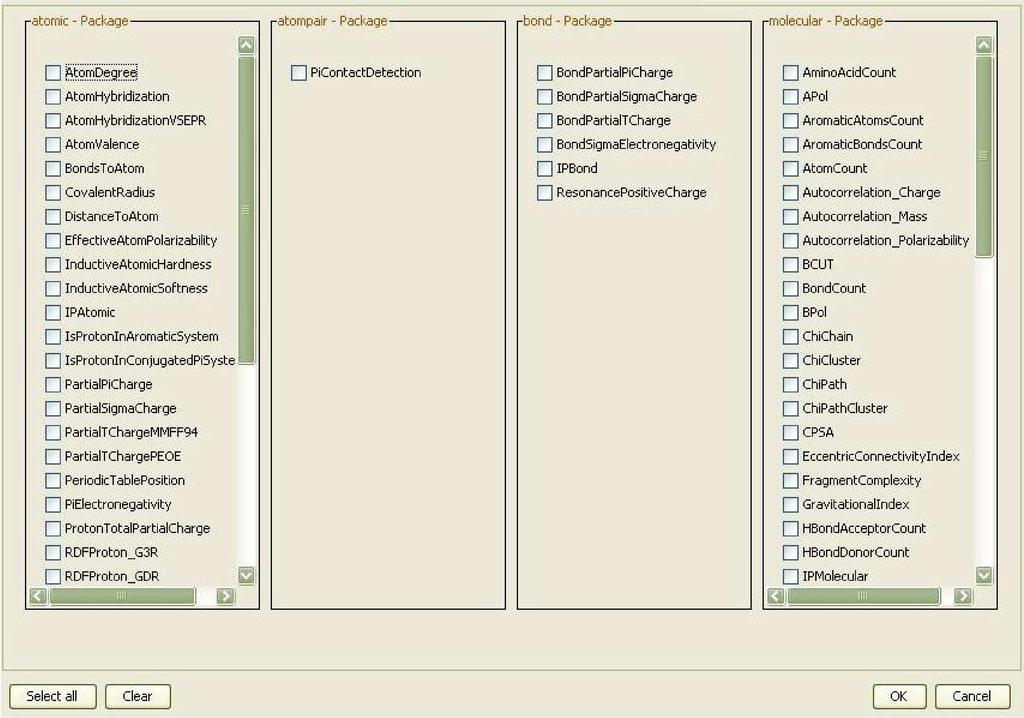
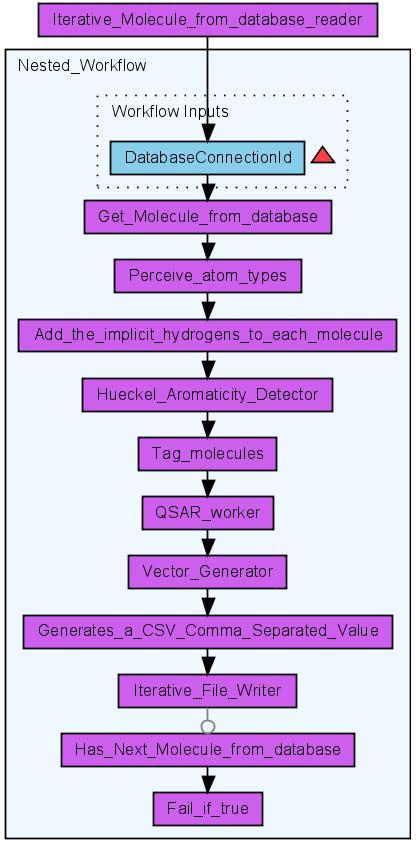
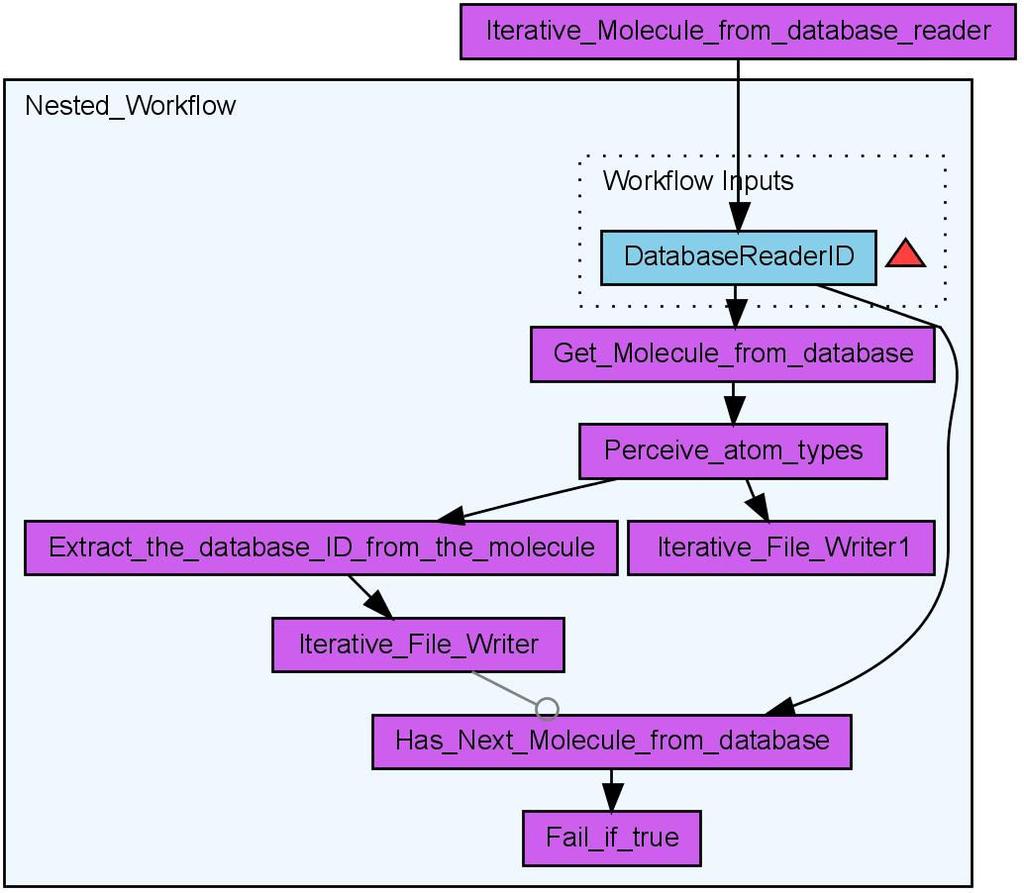
7 identifier to each molecule. This allows the identification of the corresponding Quantitative Structure-Activity Relationship (QSAR) descriptor values within the table of calculated descriptor values. The QSAR worker provides a user interface based selection of multiple QSAR descriptors (see Figure 4) for the calculation of a molecule s property vector based on the CDK. The result of the workflow is comma separated value (CSV) text file which contains the ID of the molecule and the calculate property values. The property vectors may then be used for statistical analysis, clustering or machine learning purposes. With the PostgreSQL Database back-end CDK-Taverna is able to calculate a large number of descriptors for many thousand of molecules in a reasonable time (see Figure 5). Scenario 3: Iterative Descriptor Calculation The iterative descriptor calculation workflow is a work-around which allows the treatment of hundreds of thousands of molecules. This workflow (see Figure 6) processes each molecule in the same manner as the non-iterative descriptor calculation workflow but it uses different database workers. Instead of the single database worker Get Molecules From Database three database workers are applied: Iterative Molecule From Database Reader, Get Molecule From Database and Has Next Molecule From Database. The first of the three workers is used to configure the database connection and store it within an internal object registry. The second worker gets the ID of the database connection as an input and loads molecules from the database. Only a subset of the original query is loaded using the SQL functions LIMIT and OFFSET. The last database worker checks whether the set of loaded molecules is the last of this query or if further molecules must be loaded. If the latter applies the output of this last worker would be the text value true. A last but essential worker is Fail if true. This worker throws an exception if it gets the value true as input. This worker is crucial for the nested workflow: If it fails the whole nested workflow fails. Taverna then provides a retry mechanism for a failing worker or nested workflow. This mechanism is used to re-run the nested workflow as often as necessary. This dirty workaround might become obsolete in later versions of Taverna. Scenario 4: Validation of CDK Atom Types The calculation of physiochemical properties in the Chemistry Development Kit (CDK) relies on the detection of atom types for all atoms in each molecule. The atom types describe basic atomic properties needed by the various cheminformatics algorithms implemented in the CDK. If no atom type is recognized for an atom, the atom is flagged as unknown. Based on the CDK s atom type perception functionality, we 6
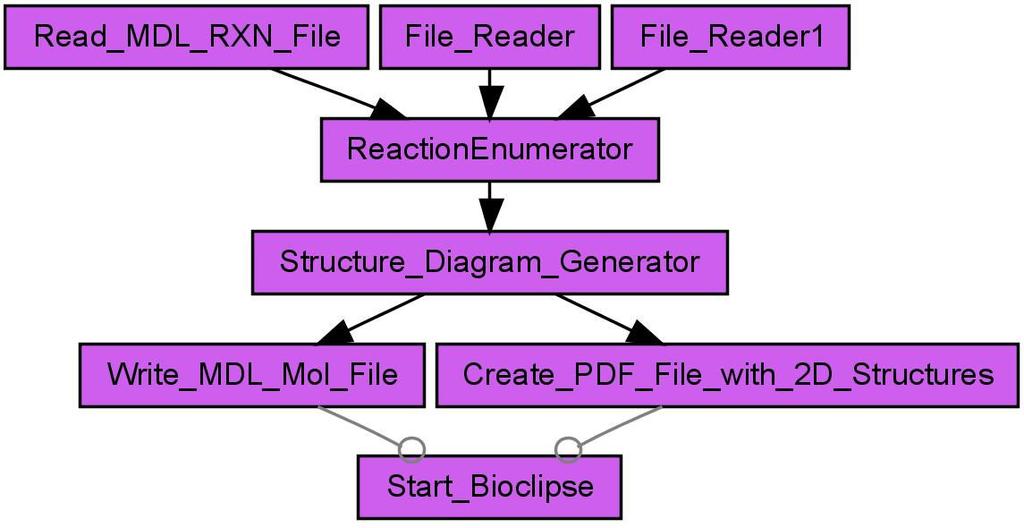
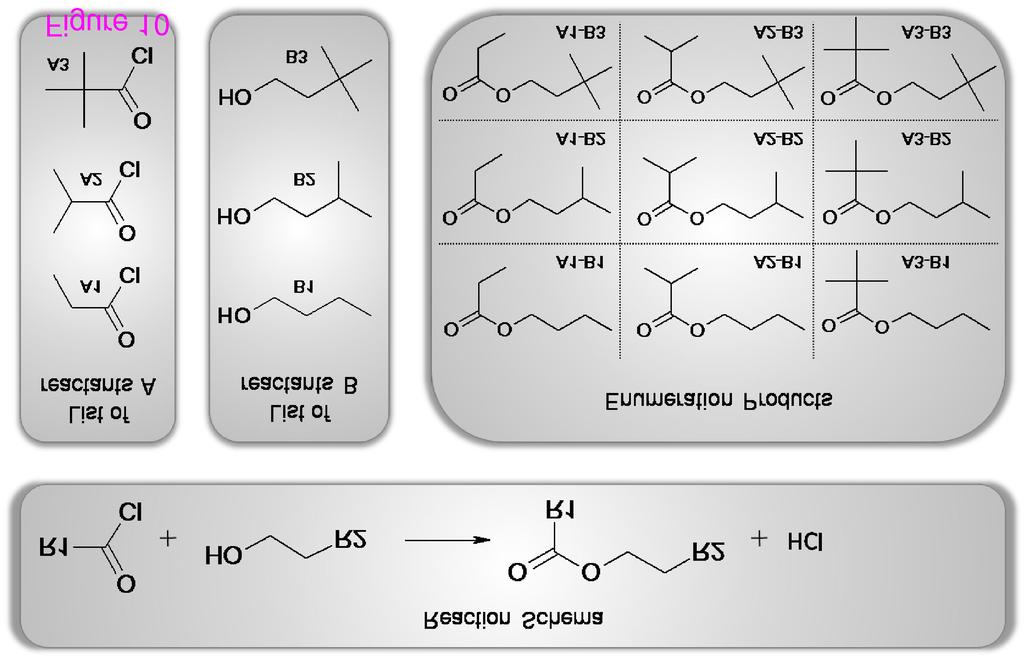
8 devised an example workflow (see Figure 7) for the validation of the CDK atom typing procedures. The detection of an unknown atom type by the CDK indicates that either the CDK lacks this specific atom type or the molecule contains chemically nonsensical atom types. In Figure 7 the Perceive atom types worker performs an atom type detection, followed by the retrieval of the database ID for those molecules with unknown atom type by the Extract the databaseid from the molecule worker. The workflow creates two text files, one containing the identifier of all molecules with unknown atom types, created by the Iterative File Writer, and a second one containing information about which atom of which molecule is unknown to the CDK. An analysis [23] of the atom type detection was performed on three different databases, two proprietary natural products databases and the open access database of Chemical Entities of Biological Interest (ChEBI) [32, 33], maintained at the European Bioinformatics Institute (EBI). The workflow was run with more than 600 thousand molecules and showed that the CDK algorithms matches the atom types quite well, but that the atom type list is not complete for metals and other heavy atoms (see Figure 8). Missing atom type definitions is a general problem to many cheminformatics algorithms and not unique to the CDK: it leads to severe problems and computation error. Therefore, initial atom type perception is an important filter for cheminformatics workflows. Scenario 5: Reaction Enumeration Markush structures are chemical drawings which represent a series of molecules by indicating locations where differences occur. These locations are marked as Heterocyclic, Alkyl, or identified by an R group, enumerating a series of possible groups, such as Methyl, Isopropyl, and Pentyl. Markush structures are commonly used in patents for describing whole compound classes and are named after Eugene A. Markush who described these kind of structures firstly in his US patent in the 1920s. In the process of reaction enumeration, Markush structures are used to design generic reactions. These reactions are usable for the enumeration of large chemical spaces, which includes the generation of chemical target libraries. The results of the enumeration has important applications in patent formulation and in High Throughput Screening (HTS). HTS experiments screen large amounts of small molecules, called molecule libraries, against one or more assays for testing for biological activity. A couple of years ago, the libraries used for a single HTS experiment consisted of up to molecules. Nowadays, more targeted libraries of a reduced size of up to molecules are used, but still commonly defined using Markush structures. For reaction enumeration, a given reaction contains different building blocks, which are needed for the 7
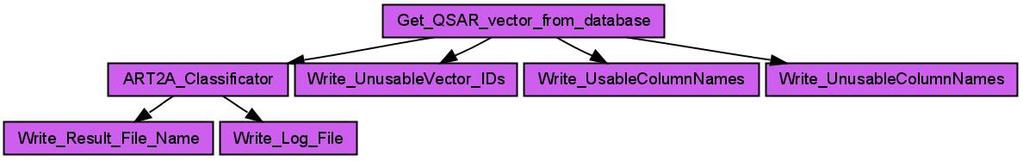
9 enumeration. Each reactant of the reaction represents a building block. A scientist then selects a number of molecules for each reactant and the reaction enumeration creates a list of all possible products. The list of products then passes a virtual screening before at last a scientist decides which products will be synthesized. Results can be visualized and inspected at the end in Bioclipse [17]. CDK-Taverna contains workers which support an enumeration task based on a generic reaction (see Figures 9 and 10). Scenario 6: Clustering Workflows During the work on this project the majority of workflows used unsupervised clustering with an implementation of the ART 2-A algorithm [34]. This algorithm was chosen because of its capability to automatically cluster open-categorical problems. Compared to traditional clustering methods like the k-means, the ART 2-A algorithm is computationally less demanding and therefore applicable especially to large data sets. Within a typical clustering workflow the Get QSAR vector from database worker loads molecule s data vectors (compare descriptor calculation workflow above) for a specific molecular SQL query from the database. This worker provides options to inspect the result vector which includes checks for values such as Not a Number (NaN) or Infinity. In addition, different thresholds may be specified for components or complete vector removal, e.g. for the removing of components whose minimum value equals its maximum value. After the loading and cleaning of a data vector, the clustering task is performed using the ART2A Classificator worker, as depicted in Figure 11. For the configuration of this worker different options are available: linear scaling of the input vector to values between 0 and 1, a switch between deterministic random and random random for the selection of the vectors to process, the definition of the convergence criteria of the clustering process, the required similarity for the convergence criteria, the maximum clustering time, and a limit for the number of clustering steps and a range for the vigilance parameter that guides the ART 2-A algorithm. The implemented ART 2-A algorithm contains two possible convergence criteria. It converges if the classification does not change after one epoch or if the scalar product of the classes between two epochs is 8
10 less than the required similarity. The clustering result is stored in form of a compressed XML document. This XML result document can be processed with different workers to create different visualizations depending on the aim of the clustering task. For chemical diversity analysis the ART 2-A worker was used for a successive top-down clustering of three chemical databases (two proprietary databases containing natural products and the ChEBI database [33] containing molecules of biological interest, see Figure 12). The occupancies of the different clusters show the similarity of the natural product databases in contrast to the ChEBI database which differs in chemical space occupation [23]. This findings will be outlined in a subsequent publication. Conclusions With CDK-Taverna we have presented the first free and open cheminformatics workflow solution for the biosciences. It allows to link and process data from various sources in visually accessible workflow diagrams without any deeper programming experience. Processing of hundreds of thousands of molecules has been demonstrated and the upper boundary is only limited by the amount of available memory. The currently implemented workers allow the processing of chemical data in various formats, provides the possibility to calculate chemical properties and allows cluster analysis of molecular descriptor vectors. The use of the PostgreSQL database with the Pgchem::tigres cheminformatics cartridge provides access to chemical databases with up to a million molecules. Availability and requirements Project name: CDK-Taverna Project home page: Operating system(s): Platform independent Programming language: Java Other requirements: Java or higher ( Taverna ( License: GNU Library or Lesser General Public License (LGPL) Any restrictions to use by non-academics: none 9
11 Authors contributions EW initiated the integration of Taverna and the CDK. CS and AZ conceived the project, and lead the further development. TK did the majority of CDK-Taverna development and developed the projects to its current state. All co-authors contributed to the manuscript. All authors read and approved the final manuscript. Acknowledgements The authors express their gratitude to the Taverna team as well as the CDK community for creating these great open tools, and like to thank Ernst-Georg Schmid for his support concerning the Pgchem::tigress functionality. References 1. The PubChem Project [ 2. Irwin J, Shoichet B: ZINC - A Free Database of Commercially Available Compounds for Virtual Screening. Journal of Chemical Information and Modeling 2005, 45: The ChEMBL Group [ 4. Williams AJ: Public chemical compound databases. Current opinion in drug discovery & development 2008, 11(3): Hassan M, Brown RD, Varma-O brien S, Rogers D: Cheminformatics analysis and learning in a data pipelining environment. Molecular diversity 2006, 10(3): Shon J, Ohkawa H, Hammer J: Scientific workflows as productivity tools for drug discovery. Current opinion in drug discovery & development 2008, 11(3): Pipeline Pilot data analysis and reporting platform [ 8. Inforsense Platform [ technology/inforsense platform/]. 9. KNIME Konstanz Information Miner [ 10. Tiwari A, Sekhar AKT: Workflow based framework for life science informatics. Computational Biology and Chemistry 2007, 31(5-6): Steinbeck C: Recent Developments in Automated Structure Elucidation of Natural Products. Natural Product Reports 2004, 21(4): Steinbeck C, Han YQ, Kuhn S, Horlacher O, Luttmann E, Willighagen E: The Chemistry Development Kit (CDK): An open-source Java library for chemo- and bioinformatics. Journal of Chemical Information and Computer Sciences 2003, 43(2): CDK-Taverna fully recognized CDK-Taverna Release on Oinn T, Addis M, Ferris J, Marvin D, Senger M, Greenwood M, Carver T, Glover K, Pocock MR, Wipat A, Li P: Taverna: a tool for the composition and enactment of bioinformatics workflows. Bioinformatics 2004, 20(17): The Open Source Definition. 10
12 17. Spjuth O, Helmus T, Willighagen EL, Kuhn S, Eklund M, Steinbeck C, Wikberg JE: Bioclipse: An open rich client workbench for chemo- and bioinformatics. BMC Bioinformatics 2007, 8(59). 18. pgchem::tigress: chemoinformatics extension to the PostgreSQL [ 19. Apache Maven [ 20. Taylor IJ, Deelman E, Gannon DB, Shields M: Workflows for e-science: Scientific Workflows for Grids. London, Springer, W3C Web Service Definition Language (WSDL) [ 22. W3C SOAP Specifications [ 23. Kuhn T: Open Source Workflow Engine for Cheminformatics: From Data Curation to Data Analysis. PhD thesis, University of Cologne myexperiment - Tags - Workflows only - cdk-taverna [ 25. PostgreSQL Database [ 26. GiST Support for PostgreSQL [ megera/postgres/gist/]. 27. Weininger D: SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. Journal of Chemical Information and Computer Sciences 1988, 28: Dalby A, Nourse JG, Hounshell WD, Gushurst AKI, Grier DL, Leland BA, Laufer J: Description of several chemical structure file formats used by computer programs developed at Molecular Design Limited. Journal of Chemical Information and Computer Sciences 1992, 32(3): Murray-Rust P, Rzepa H: Chemical Markup, XML, and the Worldwide Web. 1. Basic Principles. Journal of Chemical Information and Computer Sciences 1999, 39(6): Willighagen EL: Processing CML conventions in Java. Internet Journal of Chemistry 2001, 4: Kuhn S, Helmus T, Lancashire R, Murray-Rust P, Rzepa H, Steinbeck C, Willighagen E: Chemical Markup, XML, and the World Wide Web. 7. CMLSpect, an XML Vocabulary for Spectral Data. Journal of Chemical Information and Modeling 2007, 47(6): , 32. The database of chemical entities of biological Interest (ChEBI) [ 33. Degtyarenko K, de Matos P, Ennis M, Hastings J, Zbinden M, McNaught A, Alcantara R, Darsow M, Guedj M, Ashburner M: ChEBI: a database and ontology for chemical entities of biological interest. Nucl. Acids Res. 2008, 36: Carpenter G, Grossberg S, Rosen D: ART 2-A: an adaptive resonance algorithm for rapid categorylearning and recognition. Neural networks 1991, 4(4): myexperiment - Topological Substructure Workflow [ 36. myexperiment - Substructure Search on Database Workflow [ 37. The CDK-Taverna Blog [ time-evaluation-for-calculating-molecular-descriptors-using-the-cdk/]. 38. myexperiment - Calculation of molecular descriptors for molecules loaded from database [ 39. myexperiment - Reaction Enumeration Workflow [ 11
13 Figures [angle=0,clip=false,scale=.6]pics/substructureworkflow.pdf Figure 1: Workflow performing a topological substructure search (Scenario 1) on molecules from a MDL SDfile [35]. The input of this workflow is a SMILES string which represents the substructure. [angle=0,clip=false,scale=.2]pics/databasesubstructuresearch.png Figure 2: Workflow performing a substructure search (Scenario 1) in a database [36]. The substructure is defined with a SMILES string. The output is a PDF file with a tabular view of the molecules from the database containing the substructure. [angle=0,clip=false,scale=.5]pics/qsarworkflow.pdf Figure 3: Workflow calculating various QSAR descriptors (Scenario 2) for molecules from a PostgreSQL database. The results of the calculation are stored in a CSV file. 12
14 [angle=0,clip=true,scale=.5]pics/qsarworkeruismall.pdf Figure 4: User interface to select QSAR descriptors to be calculated for each molecule during the execution of the descriptor calculation workflow shown in Figure 3. [angle=0,clip=false,scale=.3]pics/timeneededtocalculatemoleculardescriptors.png Figure 5: Overview of the time needed to calculate different molecular descriptors for 1000 molecules [37]. [angle=0,clip=false,scale=.5]pics/iterativeqsarworkflow.png Figure 6: Workflow iteratively calculating different QSAR descriptors (Scenario 3) for molecules loaded from a PostgreSQL database [38]. The results are stored in a CSV file. [angle=0,clip=false,scale=.5]pics/atomtypingwf.pdf Figure 7: Workflow for iterative loading of molecules from a database and searches for molecules with atom types unknown to the Chemistry Development Kit (Scenario 4). [angle=0,clip=false,scale=.5]pics/atomtypingresults.png Figure 8: Allocation of the unknown atom types detected during the analysis of the ChEBI database (12367 molecules). A total of 2414 atoms in 1035 molecules (8.36%) did not have a recognized atom type. X1 summarizes unrecognized atom types for the elements Am, Cf, Cm, Dy, Es, Fm, Ga, Lr, Md, Na, Nb, No, Np, Pm, Pu, Sm, Tb, Tc, Th, and Ti. [angle=0,clip=flase,scale=.4]pics/reactionenumerationwf2.png Figure 9: Reaction enumeration (Scenario 5) loading a generic reaction from a MDL RXNfile and two reactant lists from MDL SDfiles. The products from the enumeration are stored as MDL Molfiles. Besides these files a PDF document showing the 2D structure of the products is created. At the end Bioclipse will start up to allow visualization and analysis of the results [39]. [angle=0,clip=false,scale=.5]pics/reactionenumeration.png Figure 10: Reaction enumeration example with two building blocks. For each building block, a list of three reactants is defined. This enumeration results in nine different products. [angle=0,clip=false,scale=.5]pics/atr2aclassificationwf.png Figure 11: Workflow for loading molecular descriptor data vectors from a database, followed by a ART 2-A clustering (Scenario 6). [angle=0,clip=false,scale=.5]pics/art2acluster.png Figure 12: Occupancies of six different detected clusters for two proprietary natural product databases (yellow and red) with the ChEBI database (blue), highlighting the unique character of the ChEBI database. 13
15 Tables Table 1: List of available Service Provider Interfaces that can be used to create plug-ins for Taverna to provide additional functionality. Interfaces org.embl.ebi.escience.scuflworkers.java.localworker net.sf.taverna.perspectives.perspectivespi org.embl.ebi.escience.scuflui.spi.processoractionspi org.embl.ebi.escience.scuflworkers.processorinfobean org.embl.ebi.escience.scuflui.spi.rendererspi org.embl.ebi.escience.scuflui.spi.resultmapsavespi org.embl.ebi.escience.scuflui.workbench.scavenger.spi.scavengeractionspi org.embl.ebi.escience.scuflworkers.scavengerhelper org.embl.ebi.escience.scuflui.workbench.scavenger org.embl.ebi.escience.scuflui.actions.scuflmodelactionspi org.embl.ebi.escience.scuflui.spi.uicomponentfactoryspi Table 2: The worker allocation of CDK-Taverna by function. Workers by function Number of workers Examples File I/O 15 SDFParser, CML Reader & Writer SMILES tools 2 SMILES Parser, SMILES Writer InChI parser 2 InChI Parser, InChI Generator Database I/O 7 Insert Molecules Into Database, Read Molecules From Database Molecular descriptors 42 AtomCount & LargestChain Atom descriptors 27 AtomHybridization & BondsToAtom Bond descriptors 6 PartialPiCharge Clustering 13 K-Means, ART 2-A Classification Miscellaneous 50 Substructure Search, Reaction Enumeration 14
16 Figure 1
17
18 Figure 3
19 Figure 4
20 Figure 5
21 Figure 6
22 Figure 7
23
24 Figure 9
25
26 Figure 11
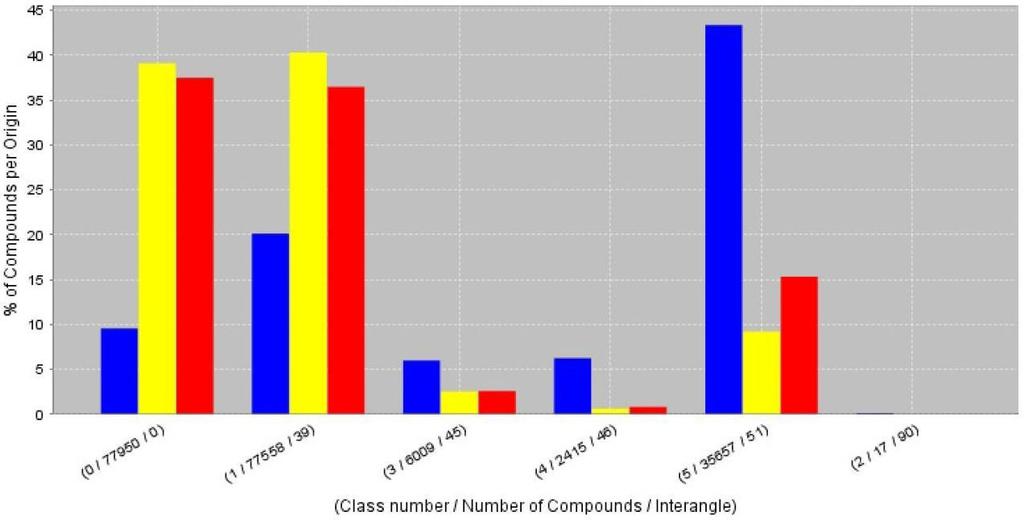
27 Figure 12
CDK & Mass Spectrometry
 CDK & Mass Spectrometry October 3, 2011 1/18 Stephan Beisken October 3, 2011 EBI is an outstation of the European Molecular Biology Laboratory. Chemistry Development Kit (CDK) An Open Source Java TM Library
CDK & Mass Spectrometry October 3, 2011 1/18 Stephan Beisken October 3, 2011 EBI is an outstation of the European Molecular Biology Laboratory. Chemistry Development Kit (CDK) An Open Source Java TM Library
Chemical Data Retrieval and Management
 Chemical Data Retrieval and Management ChEMBL, ChEBI, and the Chemistry Development Kit Stephan A. Beisken What is EMBL-EBI? Part of the European Molecular Biology Laboratory International, non-profit
Chemical Data Retrieval and Management ChEMBL, ChEBI, and the Chemistry Development Kit Stephan A. Beisken What is EMBL-EBI? Part of the European Molecular Biology Laboratory International, non-profit
Building blocks for automated elucidation of metabolites: Machine learning methods for NMR prediction
 Building blocks for automated elucidation of metabolites: Machine learning methods for NMR prediction Stefan Kuhn 1, Björn Egert 2, Steffen Neumann 2, Christoph Steinbeck 1European Bioinformatics Institute
Building blocks for automated elucidation of metabolites: Machine learning methods for NMR prediction Stefan Kuhn 1, Björn Egert 2, Steffen Neumann 2, Christoph Steinbeck 1European Bioinformatics Institute
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics
 Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
FROM MOLECULAR FORMULAS TO MARKUSH STRUCTURES
 FROM MOLECULAR FORMULAS TO MARKUSH STRUCTURES DIFFERENT LEVELS OF KNOWLEDGE REPRESENTATION IN CHEMISTRY Michael Braden, PhD ACS / San Diego/ 2016 Overview ChemAxon Who are we? Examples/use cases: Create
FROM MOLECULAR FORMULAS TO MARKUSH STRUCTURES DIFFERENT LEVELS OF KNOWLEDGE REPRESENTATION IN CHEMISTRY Michael Braden, PhD ACS / San Diego/ 2016 Overview ChemAxon Who are we? Examples/use cases: Create
New developments on the cheminformatics open workflow environment CDK-Taverna
 SOFTWARE Open Access New developments on the cheminformatics open workflow environment CDK-Taverna Andreas Truszkowski 1, Kalai Vanii Jayaseelan 2, Stefan Neumann 3, Egon L Willighagen 4, Achim Zielesny
SOFTWARE Open Access New developments on the cheminformatics open workflow environment CDK-Taverna Andreas Truszkowski 1, Kalai Vanii Jayaseelan 2, Stefan Neumann 3, Egon L Willighagen 4, Achim Zielesny
Reaxys Pipeline Pilot Components Installation and User Guide
 1 1 Reaxys Pipeline Pilot components for Pipeline Pilot 9.5 Reaxys Pipeline Pilot Components Installation and User Guide Version 1.0 2 Introduction The Reaxys and Reaxys Medicinal Chemistry Application
1 1 Reaxys Pipeline Pilot components for Pipeline Pilot 9.5 Reaxys Pipeline Pilot Components Installation and User Guide Version 1.0 2 Introduction The Reaxys and Reaxys Medicinal Chemistry Application
Practical QSAR and Library Design: Advanced tools for research teams
 DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
JOHN MAYFIELD EGON WILLIGHAGEN CHEMISTRY DEVELOPMENT KIT V2.0
 JOHN MAYFIELD EGON WILLIGHAGEN CHEMISTRY DEVELOPMENT KIT V2.0 CINF 64: ACS Fall 2017, Washington, D.C. http://cdk.github.io http://efficientbits.blogspot.com INTRODUCTION HISTORY Project started in 2000
JOHN MAYFIELD EGON WILLIGHAGEN CHEMISTRY DEVELOPMENT KIT V2.0 CINF 64: ACS Fall 2017, Washington, D.C. http://cdk.github.io http://efficientbits.blogspot.com INTRODUCTION HISTORY Project started in 2000
Rapid Application Development using InforSense Open Workflow and Daylight Technologies Deliver Discovery Value
 Rapid Application Development using InforSense Open Workflow and Daylight Technologies Deliver Discovery Value Anthony Arvanites Daylight User Group Meeting March 10, 2005 Outline 1. Company Introduction
Rapid Application Development using InforSense Open Workflow and Daylight Technologies Deliver Discovery Value Anthony Arvanites Daylight User Group Meeting March 10, 2005 Outline 1. Company Introduction
Marvin. Sketching, viewing and predicting properties with Marvin - features, tips and tricks. Gyorgy Pirok. Solutions for Cheminformatics
 Marvin Sketching, viewing and predicting properties with Marvin - features, tips and tricks Gyorgy Pirok Solutions for Cheminformatics The Marvin family The Marvin toolkit provides web-enabled components
Marvin Sketching, viewing and predicting properties with Marvin - features, tips and tricks Gyorgy Pirok Solutions for Cheminformatics The Marvin family The Marvin toolkit provides web-enabled components
Pipeline Pilot Integration
 Scientific & technical Presentation Pipeline Pilot Integration Szilárd Dóránt July 2009 The Component Collection: Quick facts Provides access to ChemAxon tools from Pipeline Pilot Free of charge Open source
Scientific & technical Presentation Pipeline Pilot Integration Szilárd Dóránt July 2009 The Component Collection: Quick facts Provides access to ChemAxon tools from Pipeline Pilot Free of charge Open source
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
Хемоінформатика. Докінг. Дизайн ліків. Біоінформатика (3 курс) Лекція 4 (частина 1)
 Лекція 4 (частина 1)") Хемоінформатика. Докінг. Дизайн ліків Біоінформатика (3 курс) Лекція 4 (частина 1) Формати файлів в хемоінформатиці Chemical information is usually provided as files or streams and many formats have been
Хемоінформатика. Докінг. Дизайн ліків Біоінформатика (3 курс) Лекція 4 (частина 1) Формати файлів в хемоінформатиці Chemical information is usually provided as files or streams and many formats have been
Ákos Tarcsay CHEMAXON SOLUTIONS
 Ákos Tarcsay CHEMAXON SOLUTIONS FINDING NOVEL COMPOUNDS WITH IMPROVED OVERALL PROPERTY PROFILES Two faces of one world Structure Footprint Linked Data Reactions Analytical Batch Phys-Chem Assay Project
Ákos Tarcsay CHEMAXON SOLUTIONS FINDING NOVEL COMPOUNDS WITH IMPROVED OVERALL PROPERTY PROFILES Two faces of one world Structure Footprint Linked Data Reactions Analytical Batch Phys-Chem Assay Project
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
JCICS Major Research Areas
 JCICS Major Research Areas Chemical Information Text Searching Structure and Substructure Searching Databases Patents George W.A. Milne C571 Lecture Fall 2002 1 JCICS Major Research Areas Chemical Computation
JCICS Major Research Areas Chemical Information Text Searching Structure and Substructure Searching Databases Patents George W.A. Milne C571 Lecture Fall 2002 1 JCICS Major Research Areas Chemical Computation
IUCLID Substance Data
 1 Workshop on CEFIC LRI Project EEM9.4 LRI AMBIT with IUCLID6 support and extended search capabilities IUCLID Substance Data Nikolay Kochev Ideaconsult Ltd. Sofia,Bulgaria 2 Chemical structure vs. Substance
1 Workshop on CEFIC LRI Project EEM9.4 LRI AMBIT with IUCLID6 support and extended search capabilities IUCLID Substance Data Nikolay Kochev Ideaconsult Ltd. Sofia,Bulgaria 2 Chemical structure vs. Substance
Introducing a Bioinformatics Similarity Search Solution
 Introducing a Bioinformatics Similarity Search Solution 1 Page About the APU 3 The APU as a Driver of Similarity Search 3 Similarity Search in Bioinformatics 3 POC: GSI Joins Forces with the Weizmann Institute
Introducing a Bioinformatics Similarity Search Solution 1 Page About the APU 3 The APU as a Driver of Similarity Search 3 Similarity Search in Bioinformatics 3 POC: GSI Joins Forces with the Weizmann Institute
Large scale classification of chemical reactions from patent data
 Large scale classification of chemical reactions from patent data Gregory Landrum NIBR Informatics, Basel Novartis Institutes for BioMedical Research 10th International Conference on Chemical Structures/
Large scale classification of chemical reactions from patent data Gregory Landrum NIBR Informatics, Basel Novartis Institutes for BioMedical Research 10th International Conference on Chemical Structures/
Capturing Chemistry. What you see is what you get In the world of mechanism and chemical transformations
 Capturing Chemistry What you see is what you get In the world of mechanism and chemical transformations Dr. Stephan Schürer ead of Intl. Sci. Content Libraria, Inc. sschurer@libraria.com Distribution of
Capturing Chemistry What you see is what you get In the world of mechanism and chemical transformations Dr. Stephan Schürer ead of Intl. Sci. Content Libraria, Inc. sschurer@libraria.com Distribution of
Analytical data, the web, and standards for unified laboratory informatics databases
 Analytical data, the web, and standards for unified laboratory informatics databases Presented By Patrick D. Wheeler & Graham A. McGibbon ACS San Diego 16 March, 2016 Sources Process, Analyze Interfaces,
Analytical data, the web, and standards for unified laboratory informatics databases Presented By Patrick D. Wheeler & Graham A. McGibbon ACS San Diego 16 March, 2016 Sources Process, Analyze Interfaces,
Improving usability and accessibility of cheminformatics tools for chemists through cyberinfrastructure and education
 Improving usability and accessibility of cheminformatics tools for chemists through cyberinfrastructure and education Rajarshi Guha, Gary D. Wiggins, David J. Wild, Mu-Hyun Baik, Marlon E. Pierce, and
Improving usability and accessibility of cheminformatics tools for chemists through cyberinfrastructure and education Rajarshi Guha, Gary D. Wiggins, David J. Wild, Mu-Hyun Baik, Marlon E. Pierce, and
Imago: open-source toolkit for 2D chemical structure image recognition
 Imago: open-source toolkit for 2D chemical structure image recognition Viktor Smolov *, Fedor Zentsev and Mikhail Rybalkin GGA Software Services LLC Abstract Different chemical databases contain molecule
Imago: open-source toolkit for 2D chemical structure image recognition Viktor Smolov *, Fedor Zentsev and Mikhail Rybalkin GGA Software Services LLC Abstract Different chemical databases contain molecule
The shortest path to chemistry data and literature
 R&D SOLUTIONS Reaxys Fact Sheet The shortest path to chemistry data and literature Designed to support the full range of chemistry research, including pharmaceutical development, environmental health &
R&D SOLUTIONS Reaxys Fact Sheet The shortest path to chemistry data and literature Designed to support the full range of chemistry research, including pharmaceutical development, environmental health &
Cross Discipline Analysis made possible with Data Pipelining. J.R. Tozer SciTegic
 Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
The Case for Use Cases
 The Case for Use Cases The integration of internal and external chemical information is a vital and complex activity for the pharmaceutical industry. David Walsh, Grail Entropix Ltd Costs of Integrating
The Case for Use Cases The integration of internal and external chemical information is a vital and complex activity for the pharmaceutical industry. David Walsh, Grail Entropix Ltd Costs of Integrating
Tautomerism in chemical information management systems
 Tautomerism in chemical information management systems Dr. Wendy A. Warr http://www.warr.com Tautomerism in chemical information management systems Author: Wendy A. Warr DOI: 10.1007/s10822-010-9338-4
Tautomerism in chemical information management systems Dr. Wendy A. Warr http://www.warr.com Tautomerism in chemical information management systems Author: Wendy A. Warr DOI: 10.1007/s10822-010-9338-4
CSD. Unlock value from crystal structure information in the CSD
 CSD CSD-System Unlock value from crystal structure information in the CSD The Cambridge Structural Database (CSD) is the world s most comprehensive and up-todate knowledge base of crystal structure data,
CSD CSD-System Unlock value from crystal structure information in the CSD The Cambridge Structural Database (CSD) is the world s most comprehensive and up-todate knowledge base of crystal structure data,
Chemical Space: Modeling Exploration & Understanding
 verview Chemical Space: Modeling Exploration & Understanding Rajarshi Guha School of Informatics Indiana University 16 th August, 2006 utline verview 1 verview 2 3 CDK R utline verview 1 verview 2 3 CDK
verview Chemical Space: Modeling Exploration & Understanding Rajarshi Guha School of Informatics Indiana University 16 th August, 2006 utline verview 1 verview 2 3 CDK R utline verview 1 verview 2 3 CDK
Navigating between patents, papers, abstracts and databases using public sources and tools
 Navigating between patents, papers, abstracts and databases using public sources and tools Christopher Southan 1 and Sean Ekins 2 TW2Informatics, Göteborg, Sweden, Collaborative Drug Discovery, North Carolina,
Navigating between patents, papers, abstracts and databases using public sources and tools Christopher Southan 1 and Sean Ekins 2 TW2Informatics, Göteborg, Sweden, Collaborative Drug Discovery, North Carolina,
Reaxys The Highlights
 Reaxys The Highlights What is Reaxys? A brand new workflow solution for research chemists and scientists from related disciplines An extensive repository of reaction and substance property data A resource
Reaxys The Highlights What is Reaxys? A brand new workflow solution for research chemists and scientists from related disciplines An extensive repository of reaction and substance property data A resource
Catching the Drift Indexing Implicit Knowledge in Chemical Digital Libraries
 Catching the Drift Indexing Implicit Knowledge in Chemical Digital Libraries Benjamin Köhncke 1, Sascha Tönnies 1, Wolf-Tilo Balke 2 1 L3S Research Center; Hannover, Germany 2 TU Braunschweig, Germany
Catching the Drift Indexing Implicit Knowledge in Chemical Digital Libraries Benjamin Köhncke 1, Sascha Tönnies 1, Wolf-Tilo Balke 2 1 L3S Research Center; Hannover, Germany 2 TU Braunschweig, Germany
Database Speaks. Ling-Kang Liu ( 劉陵崗 ) Institute of Chemistry, Academia Sinica Nangang, Taipei 115, Taiwan
 Institute of Chemistry, Academia Sinica Nangang, Taipei 115, Taiwan") Database Speaks Ling-Kang Liu ( 劉陵崗 ) Institute of Chemistry, Academia Sinica Nangang, Taipei 115, Taiwan Email: liuu@chem.sinica.edu.tw 1 OUTLINES -- Personal experiences Publication types Secondary publication
Database Speaks Ling-Kang Liu ( 劉陵崗 ) Institute of Chemistry, Academia Sinica Nangang, Taipei 115, Taiwan Email: liuu@chem.sinica.edu.tw 1 OUTLINES -- Personal experiences Publication types Secondary publication
Data Mining in the Chemical Industry. Overview of presentation
 Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Large Scale Evaluation of Chemical Structure Recognition 4 th Text Mining Symposium in Life Sciences October 10, Dr.
 Large Scale Evaluation of Chemical Structure Recognition 4 th Text Mining Symposium in Life Sciences October 10, 2006 Dr. Overview Brief introduction Chemical Structure Recognition (chemocr) Manual conversion
Large Scale Evaluation of Chemical Structure Recognition 4 th Text Mining Symposium in Life Sciences October 10, 2006 Dr. Overview Brief introduction Chemical Structure Recognition (chemocr) Manual conversion
Integrated Cheminformatics to Guide Drug Discovery
 Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
DRUG DISCOVERY TODAY ELN ELN. Chemistry. Biology. Known ligands. DBs. Generate chemistry ideas. Check chemical feasibility In-house.
 DRUG DISCOVERY TODAY Known ligands Chemistry ELN DBs Knowledge survey Therapeutic target Generate chemistry ideas Check chemical feasibility In-house Analyze SAR Synthesize or buy Report Test Journals
DRUG DISCOVERY TODAY Known ligands Chemistry ELN DBs Knowledge survey Therapeutic target Generate chemistry ideas Check chemical feasibility In-house Analyze SAR Synthesize or buy Report Test Journals
Using Web Technologies for Integrative Drug Discovery
 Using Web Technologies for Integrative Drug Discovery Qian Zhu 1 Sashikiran Challa 1 Yuying Sun 3 Michael S. Lajiness 2 David J. Wild 1 Ying Ding 3 1 School of Informatics and Computing, Indiana University,
Using Web Technologies for Integrative Drug Discovery Qian Zhu 1 Sashikiran Challa 1 Yuying Sun 3 Michael S. Lajiness 2 David J. Wild 1 Ying Ding 3 1 School of Informatics and Computing, Indiana University,
CSD. CSD-Enterprise. Access the CSD and ALL CCDC application software
 CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
Farewell, PipelinePilot Migrating the Exquiron cheminformatics platform to KNIME and the ChemAxon technology
 Farewell, PipelinePilot Migrating the Exquiron cheminformatics platform to KNIME and the ChemAxon technology Serge P. Parel, PhD ChemAxon User Group Meeting, Budapest 21 st May, 2014 Outline Exquiron Who
Farewell, PipelinePilot Migrating the Exquiron cheminformatics platform to KNIME and the ChemAxon technology Serge P. Parel, PhD ChemAxon User Group Meeting, Budapest 21 st May, 2014 Outline Exquiron Who
Chemoinformatics and information management. Peter Willett, University of Sheffield, UK
 Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
Instruction to search natural compounds on CH-NMR-NP
 Instruction to search natural compounds on CH-NMR-NP The CH-NMR-NP is a charge free service for all users. Please note that required information (name, affiliation, country, email) has to be submitted
Instruction to search natural compounds on CH-NMR-NP The CH-NMR-NP is a charge free service for all users. Please note that required information (name, affiliation, country, email) has to be submitted
Expanding the scope of literature data with document to structure tools PatentInformatics applications at Aptuit
 Expanding the scope of literature data with document to structure tools PatentInformatics applications at Aptuit Alfonso Pozzan Computational and Analytical Chemistry Drug Design and Discovery Department
Expanding the scope of literature data with document to structure tools PatentInformatics applications at Aptuit Alfonso Pozzan Computational and Analytical Chemistry Drug Design and Discovery Department
Introduction to Chemoinformatics and Drug Discovery
 Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
OECD QSAR Toolbox v.3.3. Predicting skin sensitisation potential of a chemical using skin sensitization data extracted from ECHA CHEM database
 OECD QSAR Toolbox v.3.3 Predicting skin sensitisation potential of a chemical using skin sensitization data extracted from ECHA CHEM database Outlook Background The exercise Workflow Save prediction 23.02.2015
OECD QSAR Toolbox v.3.3 Predicting skin sensitisation potential of a chemical using skin sensitization data extracted from ECHA CHEM database Outlook Background The exercise Workflow Save prediction 23.02.2015
Searching Substances in Reaxys
 Searching Substances in Reaxys Learning Objectives Understand that substances in Reaxys have different sources (e.g., Reaxys, PubChem) and can be found in Document, Reaction and Substance Records Recognize
Searching Substances in Reaxys Learning Objectives Understand that substances in Reaxys have different sources (e.g., Reaxys, PubChem) and can be found in Document, Reaction and Substance Records Recognize
Information Extraction from Chemical Images. Discovery Knowledge & Informatics April 24 th, Dr. Marc Zimmermann
 Information Extraction from Chemical Images Discovery Knowledge & Informatics April 24 th, 2006 Dr. Available Chemical Information Textbooks Reports Patents Databases Scientific journals and publications
Information Extraction from Chemical Images Discovery Knowledge & Informatics April 24 th, 2006 Dr. Available Chemical Information Textbooks Reports Patents Databases Scientific journals and publications
ADRIANA.Code and SONNIA. Tutorial
 ADRIANA.Code and SONNIA Tutorial Modeling Corticosteroid Binding Globulin Receptor Activity Molecular Networks GmbH Computerchemie July 2008 http://www.molecular-networks.com Henkestr. 91 91052 Erlangen
ADRIANA.Code and SONNIA Tutorial Modeling Corticosteroid Binding Globulin Receptor Activity Molecular Networks GmbH Computerchemie July 2008 http://www.molecular-networks.com Henkestr. 91 91052 Erlangen
EEOS 381 -Spatial Databases and GIS Applications
 EEOS 381 -Spatial Databases and GIS Applications Lecture 5 Geodatabases What is a Geodatabase? Geographic Database ESRI-coined term A standard RDBMS that stores and manages geographic data A modern object-relational
EEOS 381 -Spatial Databases and GIS Applications Lecture 5 Geodatabases What is a Geodatabase? Geographic Database ESRI-coined term A standard RDBMS that stores and manages geographic data A modern object-relational
DivCalc: A Utility for Diversity Analysis and Compound Sampling
 Molecules 2002, 7, 657-661 molecules ISSN 1420-3049 http://www.mdpi.org DivCalc: A Utility for Diversity Analysis and Compound Sampling Rajeev Gangal* SciNova Informatics, 161 Madhumanjiri Apartments,
Molecules 2002, 7, 657-661 molecules ISSN 1420-3049 http://www.mdpi.org DivCalc: A Utility for Diversity Analysis and Compound Sampling Rajeev Gangal* SciNova Informatics, 161 Madhumanjiri Apartments,
QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression
 APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor
APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor
CLRG Biocreative V
 CLRG ChemTMiner @ Biocreative V Sobha Lalitha Devi., Sindhuja Gopalan., Vijay Sundar Ram R., Malarkodi C.S., Lakshmi S., Pattabhi RK Rao Computational Linguistics Research Group, AU-KBC Research Centre
CLRG ChemTMiner @ Biocreative V Sobha Lalitha Devi., Sindhuja Gopalan., Vijay Sundar Ram R., Malarkodi C.S., Lakshmi S., Pattabhi RK Rao Computational Linguistics Research Group, AU-KBC Research Centre
Drug Informatics for Chemical Genomics...
 Drug Informatics for Chemical Genomics... An Overview First Annual ChemGen IGERT Retreat Sept 2005 Drug Informatics for Chemical Genomics... p. Topics ChemGen Informatics The ChemMine Project Library Comparison
Drug Informatics for Chemical Genomics... An Overview First Annual ChemGen IGERT Retreat Sept 2005 Drug Informatics for Chemical Genomics... p. Topics ChemGen Informatics The ChemMine Project Library Comparison
A powerful site for all chemists CHOICE CRC Handbook of Chemistry and Physics
 Chemical Databases Online A powerful site for all chemists CHOICE CRC Handbook of Chemistry and Physics Combined Chemical Dictionary Dictionary of Natural Products Dictionary of Organic Dictionary of Drugs
Chemical Databases Online A powerful site for all chemists CHOICE CRC Handbook of Chemistry and Physics Combined Chemical Dictionary Dictionary of Natural Products Dictionary of Organic Dictionary of Drugs
Chemical Journal Publishing in an Online World. Jason Wilde, Publisher Physical Sciences Nature Publishing Group ACS Spring Meeting 2009
 Chemical Journal Publishing in an Online World Jason Wilde, Publisher Physical Sciences Nature Publishing Group ACS Spring Meeting 2009 1 Overview Chemists and chemistry NPG and chemistry Nature Chemistry
Chemical Journal Publishing in an Online World Jason Wilde, Publisher Physical Sciences Nature Publishing Group ACS Spring Meeting 2009 1 Overview Chemists and chemistry NPG and chemistry Nature Chemistry
Cheminformatics Role in Pharmaceutical Industry. Randal Chen Ph.D. Abbott Laboratories Aug. 23, 2004 ACS
 Cheminformatics Role in Pharmaceutical Industry Randal Chen Ph.D. Abbott Laboratories Aug. 23, 2004 ACS Agenda The big picture for pharmaceutical industry Current technological/scientific issues Types
Cheminformatics Role in Pharmaceutical Industry Randal Chen Ph.D. Abbott Laboratories Aug. 23, 2004 ACS Agenda The big picture for pharmaceutical industry Current technological/scientific issues Types
Building innovative drug discovery alliances. Just in KNIME: Successful Process Driven Drug Discovery
 Building innovative drug discovery alliances Just in KIME: Successful Process Driven Drug Discovery Berlin KIME Spring Summit, Feb 2016 Research Informatics @ Evotec Evotec s worldwide operations 2 Pharmaceuticals
Building innovative drug discovery alliances Just in KIME: Successful Process Driven Drug Discovery Berlin KIME Spring Summit, Feb 2016 Research Informatics @ Evotec Evotec s worldwide operations 2 Pharmaceuticals
Command-line tools of ChemAxon: tips and tricks
 Command-line tools of ChemAxon: tips and tricks György Pirok Solutions for Cheminformatics Command-line interface A command-line interface (CLI) is a mechanism for interacting with a computer operating
Command-line tools of ChemAxon: tips and tricks György Pirok Solutions for Cheminformatics Command-line interface A command-line interface (CLI) is a mechanism for interacting with a computer operating
Supporting Information. Kekule.js: An Open Source JavaScript Chemoinformatics Toolkit
 Supporting Information Kekule.js: An Open Source JavaScript Chemoinformatics Toolkit Chen Jiang, *, Xi Jin, Ying Dong and Ming Chen Department of Organic Chemistry, China Pharmaceutical University, Nanjing
Supporting Information Kekule.js: An Open Source JavaScript Chemoinformatics Toolkit Chen Jiang, *, Xi Jin, Ying Dong and Ming Chen Department of Organic Chemistry, China Pharmaceutical University, Nanjing
OECD QSAR Toolbox v.4.1. Tutorial on how to predict Skin sensitization potential taking into account alert performance
 OECD QSAR Toolbox v.4.1 Tutorial on how to predict Skin sensitization potential taking into account alert performance Outlook Background Objectives Specific Aims Read across and analogue approach The exercise
OECD QSAR Toolbox v.4.1 Tutorial on how to predict Skin sensitization potential taking into account alert performance Outlook Background Objectives Specific Aims Read across and analogue approach The exercise
cheminformatics toolkits: a personal perspective
 cheminformatics toolkits: a personal perspective Roger Sayle Nextmove software ltd Cambridge uk 1 st RDKit User Group Meeting, London, UK, 4 th October 2012 overview Models of Chemistry Implicit and Explicit
cheminformatics toolkits: a personal perspective Roger Sayle Nextmove software ltd Cambridge uk 1 st RDKit User Group Meeting, London, UK, 4 th October 2012 overview Models of Chemistry Implicit and Explicit
OECD QSAR Toolbox v.3.0
 OECD QSAR Toolbox v.3.0 Step-by-step example of how to categorize an inventory by mechanistic behaviour of the chemicals which it consists Background Objectives Specific Aims Trend analysis The exercise
OECD QSAR Toolbox v.3.0 Step-by-step example of how to categorize an inventory by mechanistic behaviour of the chemicals which it consists Background Objectives Specific Aims Trend analysis The exercise
OECD QSAR Toolbox v.3.3. Step-by-step example of how to categorize an inventory by mechanistic behaviour of the chemicals which it consists
 OECD QSAR Toolbox v.3.3 Step-by-step example of how to categorize an inventory by mechanistic behaviour of the chemicals which it consists Background Objectives Specific Aims Trend analysis The exercise
OECD QSAR Toolbox v.3.3 Step-by-step example of how to categorize an inventory by mechanistic behaviour of the chemicals which it consists Background Objectives Specific Aims Trend analysis The exercise
InChI keys as standard global identifiers in chemistry web services. Russ Hillard ACS, Salt Lake City March 2009
 InChI keys as standard global identifiers in chemistry web services Russ Hillard ACS, Salt Lake City March 2009 Context of this talk We have created a web service That aggregates sources built independently
InChI keys as standard global identifiers in chemistry web services Russ Hillard ACS, Salt Lake City March 2009 Context of this talk We have created a web service That aggregates sources built independently
WEB-BASED SPATIAL DECISION SUPPORT: TECHNICAL FOUNDATIONS AND APPLICATIONS
 WEB-BASED SPATIAL DECISION SUPPORT: TECHNICAL FOUNDATIONS AND APPLICATIONS Claus Rinner University of Muenster, Germany Piotr Jankowski San Diego State University, USA Keywords: geographic information
WEB-BASED SPATIAL DECISION SUPPORT: TECHNICAL FOUNDATIONS AND APPLICATIONS Claus Rinner University of Muenster, Germany Piotr Jankowski San Diego State University, USA Keywords: geographic information
new interface and features
 Web version of SciFinder : new interface and features Bhawat Ruangying, CAS representative Updated at 22 Dec 2009 www.cas.org SciFinder web interface Technical aspects of SciFinder Web SciFinder URL :
Web version of SciFinder : new interface and features Bhawat Ruangying, CAS representative Updated at 22 Dec 2009 www.cas.org SciFinder web interface Technical aspects of SciFinder Web SciFinder URL :
Volume Editor. Hans Weghorn Faculty of Mechatronics BA-University of Cooperative Education, Stuttgart Germany
 Volume Editor Hans Weghorn Faculty of Mechatronics BA-University of Cooperative Education, Stuttgart Germany Proceedings of the 4 th Annual Meeting on Information Technology and Computer Science ITCS,
Volume Editor Hans Weghorn Faculty of Mechatronics BA-University of Cooperative Education, Stuttgart Germany Proceedings of the 4 th Annual Meeting on Information Technology and Computer Science ITCS,
has its own advantages and drawbacks, depending on the questions facing the drug discovery.
 2013 First International Conference on Artificial Intelligence, Modelling & Simulation Comparison of Similarity Coefficients for Chemical Database Retrieval Mukhsin Syuib School of Information Technology
2013 First International Conference on Artificial Intelligence, Modelling & Simulation Comparison of Similarity Coefficients for Chemical Database Retrieval Mukhsin Syuib School of Information Technology
SPATIAL INFORMATION GRID AND ITS APPLICATION IN GEOLOGICAL SURVEY
 SPATIAL INFORMATION GRID AND ITS APPLICATION IN GEOLOGICAL SURVEY K. T. He a, b, Y. Tang a, W. X. Yu a a School of Electronic Science and Engineering, National University of Defense Technology, Changsha,
SPATIAL INFORMATION GRID AND ITS APPLICATION IN GEOLOGICAL SURVEY K. T. He a, b, Y. Tang a, W. X. Yu a a School of Electronic Science and Engineering, National University of Defense Technology, Changsha,
Application integration: Providing coherent drug discovery solutions
 Application integration: Providing coherent drug discovery solutions Mitch Miller, Manish Sud, LION bioscience, American Chemical Society 22 August 2002 Overview 2 Introduction: exploring application integration
Application integration: Providing coherent drug discovery solutions Mitch Miller, Manish Sud, LION bioscience, American Chemical Society 22 August 2002 Overview 2 Introduction: exploring application integration
SciFinder Premier CAS solutions to explore all chemistry MethodsNow, PatentPak, ChemZent, SciFinder n
 ACS International Ltd vhyttinen@cas.org SciFinder Premier CAS solutions to explore all chemistry MethodsNow, PatentPak, ChemZent, SciFinder n Veli-Pekka Hyttinen May 23, 2018 CAS Hyttinen CAS supports
ACS International Ltd vhyttinen@cas.org SciFinder Premier CAS solutions to explore all chemistry MethodsNow, PatentPak, ChemZent, SciFinder n Veli-Pekka Hyttinen May 23, 2018 CAS Hyttinen CAS supports
Introduction to Chemoinformatics
 Introduction to Chemoinformatics Dr. Igor V. Tetko Helmholtz Zentrum München - German Research Center for Environmental Health (GmbH) Institute of Bioinformatics & Systems Biology (HMGU) Kyiv, 10 August
Introduction to Chemoinformatics Dr. Igor V. Tetko Helmholtz Zentrum München - German Research Center for Environmental Health (GmbH) Institute of Bioinformatics & Systems Biology (HMGU) Kyiv, 10 August
The Molecule Cloud - compact visualization of large collections of molecules
 Ertl and Rohde Journal of Cheminformatics 2012, 4:12 METHODOLOGY Open Access The Molecule Cloud - compact visualization of large collections of molecules Peter Ertl * and Bernhard Rohde Abstract Background:
Ertl and Rohde Journal of Cheminformatics 2012, 4:12 METHODOLOGY Open Access The Molecule Cloud - compact visualization of large collections of molecules Peter Ertl * and Bernhard Rohde Abstract Background:
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Zurich, February 2014 The Schrödinger extensions
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Zurich, February 2014 The Schrödinger extensions
Biology 559R: Introduction to Phylogenetic Comparative Methods Topics for this week:
 Biology 559R: Introduction to Phylogenetic Comparative Methods Topics for this week: Course general information About the course Course objectives Comparative methods: An overview R as language: uses and
Biology 559R: Introduction to Phylogenetic Comparative Methods Topics for this week: Course general information About the course Course objectives Comparative methods: An overview R as language: uses and
Reaxys Managing Complexity
 June 2009 Reaxys Managing Complexity Dr. Jürgen Swienty-Busch (j.swienty-busch@elsevier.com) What is Reaxys? Reaxys is Chemistry Covering more than 200 years of organic, organometallic and inorganic chemistry
June 2009 Reaxys Managing Complexity Dr. Jürgen Swienty-Busch (j.swienty-busch@elsevier.com) What is Reaxys? Reaxys is Chemistry Covering more than 200 years of organic, organometallic and inorganic chemistry
Overview of Geospatial Open Source Software which is Robust, Feature Rich and Standards Compliant
 Overview of Geospatial Open Source Software which is Robust, Feature Rich and Standards Compliant Cameron SHORTER, Australia Key words: Open Source Geospatial Foundation, OSGeo, Open Standards, Open Geospatial
Overview of Geospatial Open Source Software which is Robust, Feature Rich and Standards Compliant Cameron SHORTER, Australia Key words: Open Source Geospatial Foundation, OSGeo, Open Standards, Open Geospatial
Integrative Metabolic Fate Prediction and Retrosynthetic Analysis the Semantic Way
 Integrative Metabolic Fate Prediction and Retrosynthetic Analysis the Semantic Way Leonid L. Chepelev 1 and Michel Dumontier 1,2,3 1 Department of Biology, Carleton University, 1125 Colonel by Drive, Ottawa,
Integrative Metabolic Fate Prediction and Retrosynthetic Analysis the Semantic Way Leonid L. Chepelev 1 and Michel Dumontier 1,2,3 1 Department of Biology, Carleton University, 1125 Colonel by Drive, Ottawa,
Pipeline Pilot Integration
 Pipeline Pilot Integration Szilard Dorant Solutions for Cheminformatics The Component Collection: Quick facts Provides access to ChemAxon tools from Pipeline Pilot Developed and Supported by ChemAxon Free
Pipeline Pilot Integration Szilard Dorant Solutions for Cheminformatics The Component Collection: Quick facts Provides access to ChemAxon tools from Pipeline Pilot Developed and Supported by ChemAxon Free
The MDL Discovery Framework: Data and Application Integration in the Life Sciences
 MDL Information Systems Inc. San Leandro, California The MDL Discovery Framework: Data and Application Integration in the Life Sciences 2002 MDL Information Systems, Inc. All rights reserved. No part of
MDL Information Systems Inc. San Leandro, California The MDL Discovery Framework: Data and Application Integration in the Life Sciences 2002 MDL Information Systems, Inc. All rights reserved. No part of
The Changing Requirements for Informatics Systems During the Growth of a Collaborative Drug Discovery Service Company. Sally Rose BioFocus plc
 The Changing Requirements for Informatics Systems During the Growth of a Collaborative Drug Discovery Service Company Sally Rose BioFocus plc Overview History of BioFocus and acquisition of CDD Biological
The Changing Requirements for Informatics Systems During the Growth of a Collaborative Drug Discovery Service Company Sally Rose BioFocus plc Overview History of BioFocus and acquisition of CDD Biological
Applying the Semantic Web to Computational Chemistry
 Applying the Semantic Web to Computational Chemistry Neil S. Ostlund, Mirek Sopek Chemical Semantics Inc., Gainesville, Florida, USA {ostlund, sopek}@chemicalsemantics.com Abstract. Chemical Semantics
Applying the Semantic Web to Computational Chemistry Neil S. Ostlund, Mirek Sopek Chemical Semantics Inc., Gainesville, Florida, USA {ostlund, sopek}@chemicalsemantics.com Abstract. Chemical Semantics
OECD QSAR Toolbox v.3.4. Example for predicting Repeated dose toxicity of 2,3-dimethylaniline
 OECD QSAR Toolbox v.3.4 Example for predicting Repeated dose toxicity of 2,3-dimethylaniline Outlook Background Objectives The exercise Workflow Save prediction 2 Background This is a step-by-step presentation
OECD QSAR Toolbox v.3.4 Example for predicting Repeated dose toxicity of 2,3-dimethylaniline Outlook Background Objectives The exercise Workflow Save prediction 2 Background This is a step-by-step presentation
Dock Ligands from a 2D Molecule Sketch
 Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
An Integrated Approach to in-silico
 An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
Geosciences Data Digitize and Materialize, Standardization Based on Logical Inter- Domain Relationships GeoDMS
 Geosciences Data Digitize and Materialize, Standardization Based on Logical Inter- Domain Relationships GeoDMS Somayeh Veiseh Iran, Corresponding author: Geological Survey of Iran, Azadi Sq, Meraj St,
Geosciences Data Digitize and Materialize, Standardization Based on Logical Inter- Domain Relationships GeoDMS Somayeh Veiseh Iran, Corresponding author: Geological Survey of Iran, Azadi Sq, Meraj St,
OECD QSAR Toolbox v.3.3
 OECD QSAR Toolbox v.3.3 Step-by-step example on how to predict the skin sensitisation potential of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific Aims Read
OECD QSAR Toolbox v.3.3 Step-by-step example on how to predict the skin sensitisation potential of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific Aims Read
OECD QSAR Toolbox v.4.0. Tutorial on how to predict Skin sensitization potential taking into account alert performance
 OECD QSAR Toolbox v.4.0 Tutorial on how to predict Skin sensitization potential taking into account alert performance Outlook Background Objectives Specific Aims Read across and analogue approach The exercise
OECD QSAR Toolbox v.4.0 Tutorial on how to predict Skin sensitization potential taking into account alert performance Outlook Background Objectives Specific Aims Read across and analogue approach The exercise
The Electronic Representation of Chemical Structures: beyond the low hanging fruit
 The Electronic Representation of Chemical Structures: beyond the low hanging fruit How Accelrys Plans to Address the Remaining Challenges in Structure Representation and Searching: Chemically Modified
The Electronic Representation of Chemical Structures: beyond the low hanging fruit How Accelrys Plans to Address the Remaining Challenges in Structure Representation and Searching: Chemically Modified
How to Create a Substance Answer Set
 How to Create a Substance Answer Set Select among five search techniques to find substances Since substances can be described by multiple names or other characteristics, SciFinder gives you the flexibility
How to Create a Substance Answer Set Select among five search techniques to find substances Since substances can be described by multiple names or other characteristics, SciFinder gives you the flexibility
Intelligent NMR productivity tools
 Intelligent NMR productivity tools Till Kühn VP Applications Development Pittsburgh April 2016 Innovation with Integrity A week in the life of Brian Brian Works in a hypothetical pharma company / university
Intelligent NMR productivity tools Till Kühn VP Applications Development Pittsburgh April 2016 Innovation with Integrity A week in the life of Brian Brian Works in a hypothetical pharma company / university
Design and Development of a Large Scale Archaeological Information System A Pilot Study for the City of Sparti
 INTERNATIONAL SYMPOSIUM ON APPLICATION OF GEODETIC AND INFORMATION TECHNOLOGIES IN THE PHYSICAL PLANNING OF TERRITORIES Sofia, 09 10 November, 2000 Design and Development of a Large Scale Archaeological
INTERNATIONAL SYMPOSIUM ON APPLICATION OF GEODETIC AND INFORMATION TECHNOLOGIES IN THE PHYSICAL PLANNING OF TERRITORIES Sofia, 09 10 November, 2000 Design and Development of a Large Scale Archaeological
You are Building Your Organization s Geographic Knowledge
 You are Building Your Organization s Geographic Knowledge And Increasingly Making it Available Sharing Data Publishing Maps and Geo-Apps Developing Collaborative Approaches Citizens Knowledge Workers Analysts
You are Building Your Organization s Geographic Knowledge And Increasingly Making it Available Sharing Data Publishing Maps and Geo-Apps Developing Collaborative Approaches Citizens Knowledge Workers Analysts
OECD QSAR Toolbox v.3.4
 OECD QSAR Toolbox v.3.4 Step-by-step example on how to predict the skin sensitisation potential approach of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific
OECD QSAR Toolbox v.3.4 Step-by-step example on how to predict the skin sensitisation potential approach of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific
ChemDataExtractor: A toolkit for automated extraction of chemical information from the scientific literature
 : A toolkit for automated extraction of chemical information from the scientific literature Callum Court Molecular Engineering, University of Cambridge Supervisor: Dr Jacqueline Cole 1 / 20 Overview 1
: A toolkit for automated extraction of chemical information from the scientific literature Callum Court Molecular Engineering, University of Cambridge Supervisor: Dr Jacqueline Cole 1 / 20 Overview 1
GOVERNMENT GIS BUILDING BASED ON THE THEORY OF INFORMATION ARCHITECTURE
 GOVERNMENT GIS BUILDING BASED ON THE THEORY OF INFORMATION ARCHITECTURE Abstract SHI Lihong 1 LI Haiyong 1,2 LIU Jiping 1 LI Bin 1 1 Chinese Academy Surveying and Mapping, Beijing, China, 100039 2 Liaoning
GOVERNMENT GIS BUILDING BASED ON THE THEORY OF INFORMATION ARCHITECTURE Abstract SHI Lihong 1 LI Haiyong 1,2 LIU Jiping 1 LI Bin 1 1 Chinese Academy Surveying and Mapping, Beijing, China, 100039 2 Liaoning
Pascal ET is an handheld multifunction calibrator for the measurement and simulation of the following parameters: - pressure
 DATASHEET Pascal ET Pascal ET is an handheld multifunction calibrator for the measurement and simulation of the following parameters: - pressure - electrical signals (ma, mv, V, ) - temperature (TC and
DATASHEET Pascal ET Pascal ET is an handheld multifunction calibrator for the measurement and simulation of the following parameters: - pressure - electrical signals (ma, mv, V, ) - temperature (TC and
Technical Specifications. Form of the standard
 Used by popular acceptance Voluntary Implementation Mandatory Legally enforced Technical Specifications Conventions Guidelines Form of the standard Restrictive Information System Structures Contents Values
Used by popular acceptance Voluntary Implementation Mandatory Legally enforced Technical Specifications Conventions Guidelines Form of the standard Restrictive Information System Structures Contents Values
ChemmineR: A Compound Mining Toolkit for Chemical Genomics in R
 ChemmineR: A Compound Mining Toolkit for Chemical Genomics in R Yiqun Cao Anna Charisi Thomas Girke October 28, 2009 Introduction The ChemmineR package includes functions for calculating atom pair descriptors
ChemmineR: A Compound Mining Toolkit for Chemical Genomics in R Yiqun Cao Anna Charisi Thomas Girke October 28, 2009 Introduction The ChemmineR package includes functions for calculating atom pair descriptors