Analyzing six types of protein-protein interfaces. Yanay Ofran and Burkhard Rost
|
|
|
- Percival Douglas
- 6 years ago
- Views:
Transcription
1 Analyzing six types of protein-protein interfaces Yanay Ofran and Burkhard Rost
2 Goal of the paper To check 1. If there is significant difference in amino acid composition in various interfaces of protein-protein interactions 2. If there is preference b/w pairs of residues in each of the interfaces
3 Difference in data with previous studies Different types of interactions are just classified as internal and external NO DIFFERECE B/W HOMOMULTIMERS AND HETERO-MULTIMERS NO DIFFERENCE B/W PERMANENT INTERACTIONS AND TRANCIENT INTERACTIONS Different types of interactions are classified into six categories Hand selected Automated Patches Contacts
4 WHY DIFFERENCIATE B/W Patches and Contacts? Such surface patches may not capture all aspects of protein interactions. For example, slightly buried residues with long sidechains may be missed, although they participate in interfaces. Some residues identified as part of a surface patch may form important contacts, while others may not form contacts at all. Therefore, the analysis of surface patches may not capture all residue residue contacts that underlie the interaction.
5 Advantage of automated way to differentiate multimers There is no simple way to distinguish automatically between (i) interfaces between two chains that belong to one multi-chain protein and (ii) interfaces between two different proteins.
6 Data set: Largest possible non-redundant subset of PDB: no pair of proteins in that set had more than 25% identity over 100 aligned residues The non-redundant set included 1812 high-resolution structures. We excluded NMR structures, theoretical models, and chains shorter than 30 residues. Of these proteins, 936 (51%)had resolutions below 2A, 74 proteins (4%) had resolutions above 3A.
7 Generation of dataset Identifying interacting residues from complexes in PDB We defined a residue pair to be in contact if the distance between the closest of their respective atoms was 6 A and their sequence separation was three or more residues. Previous studies used distances between 4A and 12A between two Cα or Cβ atoms.
8 Data set statistics Type of interface Number of contacts Internal intra-domain 1 3,340,485 domain-domain 2 255,144 External homo-obligomers 3 218,104 homo-complexes 4 3,077 hetero-obligomers 5 18,886 hetero-complexes 6 166,412
9 Data mining To differentiate b/w 1. Homo-multimers & heteromultimers more than 10% difference in sequence 2. Permenent homo-multimers & transient homomultimers PDB & DIP - annotated as functional monomer 3. Permenent Hetero multimers & transient Hetero multimers cross reference to SWISSSPROT- if different chain in PDB included in same swissprot file it is permanent complex

10 Amino acids in interfaces Amino acid composition of six interface types. The propensities of all residues found in SWISS-PROT were used as background. If the frequency of an amino acid is similar to its frequency in SWISS-PROT, the height of the bar is close to zero. Over-representation results in a positive bar, and under-representation results in a negative bar. Ofran, Rost, J. Mol. Biol. 325, 377 (2003)
11 Whats wrong with Standard test of significance? Problematic when the data sets are small Another problem with these tests that is not commonly noted, is that the application of significance tests to very large data sets with a few degrees of freedom, like those analyzed here, can lead to severe inferential mistakes
12 sample P JS?... sample Q1 sample Q2 sample Qn... data set 1 data set 2 data set n
13 Amino acid composition 1. Pick a random sample P with 1000 residues from one of the six sets of residue residue contacts. 2. Pick six random samples Q1 Q6 with 1000 residues from each of the six sets. 3. Find the set Qp from Q1 Q6 least divergent from set P by measuring the JS divergence between P and Q1 Q6. 4. Record the types of interactions from which P and QP were sampled. We repeated this procedure 6000 times (1000 for each of the six types of interactions). If the residue composition differed significantly between the types, we expect Qp to be, in most cases, the sample that was sampled from the same population of P. That is, we expect that in most cases, P and the sample most similar to P were sampled from contacts of the same interface type.
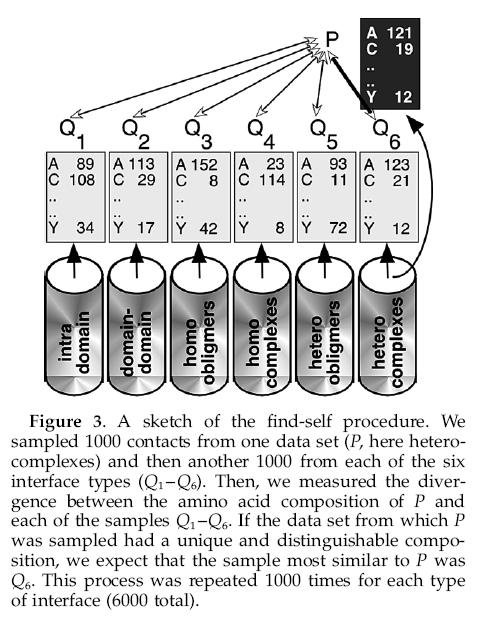
14
15 Measuring the divergence between distributions JS (p 1,p 2 ) = H (π 1 p 1 + π 2 p 2 ) - π 1 H (p 1 ) - π 2 H (p 2 ) with: π 1 and π 2 >= 0, and π 1 + π 2 =1 and H(p) =- Σ i p(x i )log 2 (x i ) P1 and P2 are prior probabilities for distributions p1 and p2 Lin (1991) IEEE Transactions on Information Theory 37:
16 Significant difference between amino acid composition of six interface types internal external intradomain domaindomain homoobligomer homocomplex heteroobligomer heterocomplex intra-domain domain-domain Homo-obligomer Homo-complex Hetero-obligomer Hetero-complex
17 Significant difference in composition of six interfaces and surface residues internal intradomain domaindomain homoobligomer homocomplex heteroobligome r heterocomplex Surface exposed intra-domain domain-domain Homo-obligomer Homo-complex Hetero-obligomer Hetero-complex Exposed 100
18 Significant difference b/w contact preferences for six interface types internal external intradomain domaindomain homoobligomer homocomplex heteroobligomer heterocomplex intra-domain domain-domain Homo-obligomer Homo-complex Hetero-obligomer Hetero-complex
19 CONCLUSIONS ON STUDY OF RESIDUE CONTACT PREFERENCES Homo-complexes depleted in salt-bridges and rich in contacts between identical residues: Hydrophobic hydrophilic interactions dominated intra-domain, domain domain and heterocomplex interfaces Cysteine bridges were observed more often than expected for all interface types. Salt bridges were common with the exception of homocomplexes, for which they were observed less often than expected. Homo-complexes exhibited an extreme general preference for interactions between identical amino acid residues. Furthermore, overall the contact preferences also stood out most for homo-complexes.
20 Measuring residue residue preferences After it had been established that the amino acid composition differed between the six interface types, the six lists was to compute the likelihood of forming contacts between each pair of amino acid residues. In particular, the log odds ratio of the observed frequency of the pair over its expected frequency is compiled
21 Lx(i,j) = log 2 (P(i,j)/(P(i)P(j))) where the subscript x represents one of the six types of interfaces (intra-domain, domain domain, homoobligomers, homo-complex, hetero-obligomers and hetero-complex), i and j are types of amino acid residue, P(i,j ) is the probability of a contact between amino acids of type i and j in interfaces of type x, and P(i ) and P( j ) are the probability of occurrence for amino acid residues i and j, respectively, in the interfaces of type x. Hence, the denominator describes the probability of a contact between i and j if the formation of contacts between i and j were random. On the basis of this equation, we generated six matrices for the likelihood for all possible contacts in each interface type.
 Intra-domain: hydrophobic core is clear (B) domain domain, (C) obligatory homooligomers (homo-obligomers), (D) transient homo-oligomers (homo-complexes), (E) obligatory hetero-oligomers")
22 Pairing frequencies at interfaces Residue residue preferences. (A) Intra-domain: hydrophobic core is clear (B) domain domain, (C) obligatory homooligomers (homo-obligomers), (D) transient homo-oligomers (homo-complexes), (E) obligatory hetero-oligomers (heteroobligomers), and (F) transient heterooligomers (hetero-complexes). A red square indicates that the interaction occurs more frequently than expected; a blue square indicates that it occurs less frequently than expected. The amino acid residues are ordered according to hydrophobicity, with isoleucine as the most hydrophobic and arginine as the least hydrophobic. Ofran, Rost, J. Mol. Biol. 325, 377 (2003)
23 Observing the Result For the other interface types, we observed strong self-interaction preferences exclusively for cysteine residues, which are known to stabilise interactions through forming cysteine bridges Confirming earlier findings, we found saltbridges abundant in interfaces
24 Observing the Result Based on amino acid composition, some studies hypothesize that hydrophobic interactions are more frequent in permanent interactions than in transient interactions. This hypothesis is confirmed by our residue residue preference data, both for homo-obligomers and for hetero obligomers
25 Novel components that yielded results Set of interfaces were data-mined from PDB which is far larger than datasets analyzed before Analysis on a more finely grained distinction of interfaces than explored previously. Distinguished b/w two types of internal interactions Distinguished b/w four types of external interactions analyzed interface contacts rather than surface patches Established the statistical significance of differences through a rigorous information theory derived procedure.
26 Critical analysis Criteria for differentiating different types of complexes - no validation dependent on annotation if DIP
27 CONCLUSIONS Six types of interfaces analysed differed in amino acid composition residue contact preferences
28 Discussion/questions?
Measuring quaternary structure similarity using global versus local measures.
 Supplementary Figure 1 Measuring quaternary structure similarity using global versus local measures. (a) Structural similarity of two protein complexes can be inferred from a global superposition, which
Supplementary Figure 1 Measuring quaternary structure similarity using global versus local measures. (a) Structural similarity of two protein complexes can be inferred from a global superposition, which
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Characterization of Protein Protein Interfaces
 Protein J DOI 1.17/s193-7-91-x Characterization of Protein Protein Changhui Yan Æ Feihong Wu Æ Robert L. Jernigan Æ Drena Dobbs Æ Vasant Honavar Ó Springer Science+Business Media, LLC 7 Abstract We analyze
Protein J DOI 1.17/s193-7-91-x Characterization of Protein Protein Changhui Yan Æ Feihong Wu Æ Robert L. Jernigan Æ Drena Dobbs Æ Vasant Honavar Ó Springer Science+Business Media, LLC 7 Abstract We analyze
Analysis of Relevant Physicochemical Properties in Obligate and Non-obligate Protein-protein Interactions
 Analysis of Relevant Physicochemical Properties in Obligate and Non-obligate Protein-protein Interactions Mina Maleki, Md. Mominul Aziz, Luis Rueda School of Computer Science, University of Windsor 401
Analysis of Relevant Physicochemical Properties in Obligate and Non-obligate Protein-protein Interactions Mina Maleki, Md. Mominul Aziz, Luis Rueda School of Computer Science, University of Windsor 401
From Amino Acids to Proteins - in 4 Easy Steps
 From Amino Acids to Proteins - in 4 Easy Steps Although protein structure appears to be overwhelmingly complex, you can provide your students with a basic understanding of how proteins fold by focusing
From Amino Acids to Proteins - in 4 Easy Steps Although protein structure appears to be overwhelmingly complex, you can provide your students with a basic understanding of how proteins fold by focusing
Protein-protein Interaction Prediction using Desolvation Energies and Interface Properties
 200 IEEE International Conference on Bioinformatics and Biomedicine Protein-protein Interaction Prediction using Desolvation Energies and Interface Properties Luis Rueda, Sridip Banerjee, Md. Mominul Aziz,
200 IEEE International Conference on Bioinformatics and Biomedicine Protein-protein Interaction Prediction using Desolvation Energies and Interface Properties Luis Rueda, Sridip Banerjee, Md. Mominul Aziz,
PDBe TUTORIAL. PDBePISA (Protein Interfaces, Surfaces and Assemblies)
") PDBe TUTORIAL PDBePISA (Protein Interfaces, Surfaces and Assemblies) http://pdbe.org/pisa/ This tutorial introduces the PDBePISA (PISA for short) service, which is a webbased interactive tool offered by
PDBe TUTORIAL PDBePISA (Protein Interfaces, Surfaces and Assemblies) http://pdbe.org/pisa/ This tutorial introduces the PDBePISA (PISA for short) service, which is a webbased interactive tool offered by
SUPPLEMENTARY MATERIALS
 SUPPLEMENTARY MATERIALS Enhanced Recognition of Transmembrane Protein Domains with Prediction-based Structural Profiles Baoqiang Cao, Aleksey Porollo, Rafal Adamczak, Mark Jarrell and Jaroslaw Meller Contact:
SUPPLEMENTARY MATERIALS Enhanced Recognition of Transmembrane Protein Domains with Prediction-based Structural Profiles Baoqiang Cao, Aleksey Porollo, Rafal Adamczak, Mark Jarrell and Jaroslaw Meller Contact:
Protein Secondary Structure Prediction
 part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 the goal is the prediction of the secondary structure conformation which is local each amino
part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 the goal is the prediction of the secondary structure conformation which is local each amino
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Motif Prediction in Amino Acid Interaction Networks
 Motif Prediction in Amino Acid Interaction Networks Omar GACI and Stefan BALEV Abstract In this paper we represent a protein as a graph where the vertices are amino acids and the edges are interactions
Motif Prediction in Amino Acid Interaction Networks Omar GACI and Stefan BALEV Abstract In this paper we represent a protein as a graph where the vertices are amino acids and the edges are interactions
Can protein model accuracy be. identified? NO! CBS, BioCentrum, Morten Nielsen, DTU
 Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Packing of Secondary Structures
 7.88 Lecture Notes - 4 7.24/7.88J/5.48J The Protein Folding and Human Disease Professor Gossard Retrieving, Viewing Protein Structures from the Protein Data Base Helix helix packing Packing of Secondary
7.88 Lecture Notes - 4 7.24/7.88J/5.48J The Protein Folding and Human Disease Professor Gossard Retrieving, Viewing Protein Structures from the Protein Data Base Helix helix packing Packing of Secondary
Structure and evolution of the spliceosomal peptidyl-prolyl cistrans isomerase Cwc27
 Acta Cryst. (2014). D70, doi:10.1107/s1399004714021695 Supporting information Volume 70 (2014) Supporting information for article: Structure and evolution of the spliceosomal peptidyl-prolyl cistrans isomerase
Acta Cryst. (2014). D70, doi:10.1107/s1399004714021695 Supporting information Volume 70 (2014) Supporting information for article: Structure and evolution of the spliceosomal peptidyl-prolyl cistrans isomerase
1-D Predictions. Prediction of local features: Secondary structure & surface exposure
 1-D Predictions Prediction of local features: Secondary structure & surface exposure 1 Learning Objectives After today s session you should be able to: Explain the meaning and usage of the following local
1-D Predictions Prediction of local features: Secondary structure & surface exposure 1 Learning Objectives After today s session you should be able to: Explain the meaning and usage of the following local
Bi 8 Midterm Review. TAs: Sarah Cohen, Doo Young Lee, Erin Isaza, and Courtney Chen
 Bi 8 Midterm Review TAs: Sarah Cohen, Doo Young Lee, Erin Isaza, and Courtney Chen The Central Dogma Biology Fundamental! Prokaryotes and Eukaryotes Nucleic Acid Components Nucleic Acid Structure DNA Base
Bi 8 Midterm Review TAs: Sarah Cohen, Doo Young Lee, Erin Isaza, and Courtney Chen The Central Dogma Biology Fundamental! Prokaryotes and Eukaryotes Nucleic Acid Components Nucleic Acid Structure DNA Base
Protein structure. Protein structure. Amino acid residue. Cell communication channel. Bioinformatics Methods
 Cell communication channel Bioinformatics Methods Iosif Vaisman Email: ivaisman@gmu.edu SEQUENCE STRUCTURE DNA Sequence Protein Sequence Protein Structure Protein structure ATGAAATTTGGAAACTTCCTTCTCACTTATCAGCCACCT...
Cell communication channel Bioinformatics Methods Iosif Vaisman Email: ivaisman@gmu.edu SEQUENCE STRUCTURE DNA Sequence Protein Sequence Protein Structure Protein structure ATGAAATTTGGAAACTTCCTTCTCACTTATCAGCCACCT...
ORGANIC - BROWN 8E CH AMINO ACIDS AND PROTEINS.
 !! www.clutchprep.com CONCEPT: INTRODUCTION TO PROTEINS Proteins are polypeptides that have some biological function. Peptides are composed of polymers of monomeric units called α-amino acids The 20 most
!! www.clutchprep.com CONCEPT: INTRODUCTION TO PROTEINS Proteins are polypeptides that have some biological function. Peptides are composed of polymers of monomeric units called α-amino acids The 20 most
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
Protein Secondary Structure Prediction
 Protein Secondary Structure Prediction Doug Brutlag & Scott C. Schmidler Overview Goals and problem definition Existing approaches Classic methods Recent successful approaches Evaluating prediction algorithms
Protein Secondary Structure Prediction Doug Brutlag & Scott C. Schmidler Overview Goals and problem definition Existing approaches Classic methods Recent successful approaches Evaluating prediction algorithms
Supplementary text for the section Interactions conserved across species: can one select the conserved interactions?
 1 Supporting Information: What Evidence is There for the Homology of Protein-Protein Interactions? Anna C. F. Lewis, Nick S. Jones, Mason A. Porter, Charlotte M. Deane Supplementary text for the section
1 Supporting Information: What Evidence is There for the Homology of Protein-Protein Interactions? Anna C. F. Lewis, Nick S. Jones, Mason A. Porter, Charlotte M. Deane Supplementary text for the section
Lec.1 Chemistry Of Water
 Lec.1 Chemistry Of Water Biochemistry & Medicine Biochemistry can be defined as the science concerned with the chemical basis of life. Biochemistry can be described as the science concerned with the chemical
Lec.1 Chemistry Of Water Biochemistry & Medicine Biochemistry can be defined as the science concerned with the chemical basis of life. Biochemistry can be described as the science concerned with the chemical
Useful background reading
 Overview of lecture * General comment on peptide bond * Discussion of backbone dihedral angles * Discussion of Ramachandran plots * Description of helix types. * Description of structures * NMR patterns
Overview of lecture * General comment on peptide bond * Discussion of backbone dihedral angles * Discussion of Ramachandran plots * Description of helix types. * Description of structures * NMR patterns
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CISC 636 Computational Biology & Bioinformatics (Fall 2016)
") CISC 636 Computational Biology & Bioinformatics (Fall 2016) Predicting Protein-Protein Interactions CISC636, F16, Lec22, Liao 1 Background Proteins do not function as isolated entities. Protein-Protein
CISC 636 Computational Biology & Bioinformatics (Fall 2016) Predicting Protein-Protein Interactions CISC636, F16, Lec22, Liao 1 Background Proteins do not function as isolated entities. Protein-Protein
Structure, mechanism and ensemble formation of the Alkylhydroperoxide Reductase subunits. AhpC and AhpF from Escherichia coli
 Structure, mechanism and ensemble formation of the Alkylhydroperoxide Reductase subunits AhpC and AhpF from Escherichia coli Phat Vinh Dip 1,#, Neelagandan Kamariah 2,#, Malathy Sony Subramanian Manimekalai
Structure, mechanism and ensemble formation of the Alkylhydroperoxide Reductase subunits AhpC and AhpF from Escherichia coli Phat Vinh Dip 1,#, Neelagandan Kamariah 2,#, Malathy Sony Subramanian Manimekalai
The prediction of membrane protein types with NPE
 The prediction of membrane protein types with NPE Lipeng Wang 1a), Zhanting Yuan 1, Xuhui Chen 1, and Zhifang Zhou 2 1 College of Electrical and Information Engineering Lanzhou University of Technology,
The prediction of membrane protein types with NPE Lipeng Wang 1a), Zhanting Yuan 1, Xuhui Chen 1, and Zhifang Zhou 2 1 College of Electrical and Information Engineering Lanzhou University of Technology,
Protein Structure Prediction
 Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Insights into Protein Protein Interfaces using a Bayesian Network Prediction Method
 doi:10.1016/j.jmb.2006.07.028 J. Mol. Biol. (2006) 362, 365 386 Insights into Protein Protein Interfaces using a Bayesian Network Prediction Method James R. Bradford 1, Chris J. Needham 2, Andrew J. Bulpitt
doi:10.1016/j.jmb.2006.07.028 J. Mol. Biol. (2006) 362, 365 386 Insights into Protein Protein Interfaces using a Bayesian Network Prediction Method James R. Bradford 1, Chris J. Needham 2, Andrew J. Bulpitt
Protein Structure. Hierarchy of Protein Structure. Tertiary structure. independently stable structural unit. includes disulfide bonds
 Protein Structure Hierarchy of Protein Structure 2 3 Structural element Primary structure Secondary structure Super-secondary structure Domain Tertiary structure Quaternary structure Description amino
Protein Structure Hierarchy of Protein Structure 2 3 Structural element Primary structure Secondary structure Super-secondary structure Domain Tertiary structure Quaternary structure Description amino
PREDICTION OF PROTEIN BINDING SITES BY COMBINING SEVERAL METHODS
 PREDICTION OF PROTEIN BINDING SITES BY COMBINING SEVERAL METHODS T. Z. SEN, A. KLOCZKOWSKI, R. L. JERNIGAN L.H. Baker Center for Bioinformatics and Biological Statistics, Iowa State University Ames, IA
PREDICTION OF PROTEIN BINDING SITES BY COMBINING SEVERAL METHODS T. Z. SEN, A. KLOCZKOWSKI, R. L. JERNIGAN L.H. Baker Center for Bioinformatics and Biological Statistics, Iowa State University Ames, IA
Acta Cryst. (2017). D73, doi: /s
. D73, doi: /s") Acta Cryst. (2017). D73, doi:10.1107/s2059798317010932 Supporting information Volume 73 (2017) Supporting information for article: Designing better diffracting crystals of biotin carboxyl carrier protein
Acta Cryst. (2017). D73, doi:10.1107/s2059798317010932 Supporting information Volume 73 (2017) Supporting information for article: Designing better diffracting crystals of biotin carboxyl carrier protein
Protein Structures. 11/19/2002 Lecture 24 1
 Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Bioinformatics: Secondary Structure Prediction
 Bioinformatics: Secondary Structure Prediction Prof. David Jones d.t.jones@ucl.ac.uk Possibly the greatest unsolved problem in molecular biology: The Protein Folding Problem MWMPPRPEEVARK LRRLGFVERMAKG
Bioinformatics: Secondary Structure Prediction Prof. David Jones d.t.jones@ucl.ac.uk Possibly the greatest unsolved problem in molecular biology: The Protein Folding Problem MWMPPRPEEVARK LRRLGFVERMAKG
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics
 Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Using Information Theory to Reduce Complexities of Neural Networks in Protein Secondary Structure Prediction
 Boon Thye Thomas Yeo 4662474 Biochem 218 Final Project Using Information Theory to Reduce Complexities of Neural Networks in Protein Secondary Structure Prediction 1 Abstract Neural networks are commonly
Boon Thye Thomas Yeo 4662474 Biochem 218 Final Project Using Information Theory to Reduce Complexities of Neural Networks in Protein Secondary Structure Prediction 1 Abstract Neural networks are commonly
ARE BINDING RESIDUES CONSERVED?
 ARE BINDING RESIDUES CONSERVED? CHRISTOS OUZOUNIS 1, CAROL PEREZ-IRRATXETA2, CHRIS SANDER1, ALFONSO VALENCIA2,c 1The European Bioinfonnatics Institute, EMBL Outstation, Cambridge UK 2Protein Design Group,
ARE BINDING RESIDUES CONSERVED? CHRISTOS OUZOUNIS 1, CAROL PEREZ-IRRATXETA2, CHRIS SANDER1, ALFONSO VALENCIA2,c 1The European Bioinfonnatics Institute, EMBL Outstation, Cambridge UK 2Protein Design Group,
BIOINFORMATICS: An Introduction
 BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
Analysis of Molecular Recognition Features (MoRFs)
") doi:10.1016/j.jmb.2006.07.087 J. Mol. Biol. (2006) 362, 1043 1059 Analysis of Molecular Recognition Features (MoRFs) Amrita Mohan 1, Christopher J. Oldfield 1,2, Predrag Radivojac 1 Vladimir Vacic 2, Marc
doi:10.1016/j.jmb.2006.07.087 J. Mol. Biol. (2006) 362, 1043 1059 Analysis of Molecular Recognition Features (MoRFs) Amrita Mohan 1, Christopher J. Oldfield 1,2, Predrag Radivojac 1 Vladimir Vacic 2, Marc
Statistical Analysis and Prediction of Protein Protein Interfaces
 PROTEINS: Structure, Function, and Bioinformatics 60:353 366 (2005) Statistical Analysis and Prediction of Protein Protein Interfaces Andrew J. Bordner* and Ruben Abagyan Molsoft LLC, San Diego, California
PROTEINS: Structure, Function, and Bioinformatics 60:353 366 (2005) Statistical Analysis and Prediction of Protein Protein Interfaces Andrew J. Bordner* and Ruben Abagyan Molsoft LLC, San Diego, California
QUANTA Protein Design MAY 2006
 QUANTA 2006 Protein Design MAY 2006 Copyright (1) Copyright Copyright 2006, Accelrys Software Inc. All rights reserved. The Accelrys name and logo are registered trademarks of Accelrys Software Inc. This
QUANTA 2006 Protein Design MAY 2006 Copyright (1) Copyright Copyright 2006, Accelrys Software Inc. All rights reserved. The Accelrys name and logo are registered trademarks of Accelrys Software Inc. This
Lecture 4 Model Answers to Problems
 Self-Study Problems / Exam Preparation Construct and annotate a valence MO diagram for H 2 CN -. Use your diagram to explain why the neutral radical is more stable than the anion. (this is an old exam
Self-Study Problems / Exam Preparation Construct and annotate a valence MO diagram for H 2 CN -. Use your diagram to explain why the neutral radical is more stable than the anion. (this is an old exam
Modeling for 3D structure prediction
 Modeling for 3D structure prediction What is a predicted structure? A structure that is constructed using as the sole source of information data obtained from computer based data-mining. However, mixing
Modeling for 3D structure prediction What is a predicted structure? A structure that is constructed using as the sole source of information data obtained from computer based data-mining. However, mixing
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Optimization and Frustration:
 Optimization and Frustration: The Dynamical Lattice Model of Proteins Sigismund Kobe and Frank Dressel Institut für Theoretische Physik, Technische Universität Dresden, D 01062 Dresden, Germany (http://www.physik.tu-dresden.de/~fdressel/dlm-main.html)
Optimization and Frustration: The Dynamical Lattice Model of Proteins Sigismund Kobe and Frank Dressel Institut für Theoretische Physik, Technische Universität Dresden, D 01062 Dresden, Germany (http://www.physik.tu-dresden.de/~fdressel/dlm-main.html)
Annotation Error in Public Databases ALEXANDRA SCHNOES UNIVERSITY OF CALIFORNIA, SAN FRANCISCO OCTOBER 25, 2010
 Annotation Error in Public Databases ALEXANDRA SCHNOES UNIVERSITY OF CALIFORNIA, SAN FRANCISCO OCTOBER 25, 2010 1 New genomes (and metagenomes) sequenced every day... 2 3 3 3 3 3 3 3 3 3 Computational
Annotation Error in Public Databases ALEXANDRA SCHNOES UNIVERSITY OF CALIFORNIA, SAN FRANCISCO OCTOBER 25, 2010 1 New genomes (and metagenomes) sequenced every day... 2 3 3 3 3 3 3 3 3 3 Computational
Sequence comparison: Score matrices. Genome 559: Introduction to Statistical and Computational Genomics Prof. James H. Thomas
 Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas Informal inductive proof of best alignment path onsider the last step in the best
Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas Informal inductive proof of best alignment path onsider the last step in the best
Hands-On Nine The PAX6 Gene and Protein
 Hands-On Nine The PAX6 Gene and Protein Main Purpose of Hands-On Activity: Using bioinformatics tools to examine the sequences, homology, and disease relevance of the Pax6: a master gene of eye formation.
Hands-On Nine The PAX6 Gene and Protein Main Purpose of Hands-On Activity: Using bioinformatics tools to examine the sequences, homology, and disease relevance of the Pax6: a master gene of eye formation.
Automatic Epitope Recognition in Proteins Oriented to the System for Macromolecular Interaction Assessment MIAX
 Genome Informatics 12: 113 122 (2001) 113 Automatic Epitope Recognition in Proteins Oriented to the System for Macromolecular Interaction Assessment MIAX Atsushi Yoshimori Carlos A. Del Carpio yosimori@translell.eco.tut.ac.jp
Genome Informatics 12: 113 122 (2001) 113 Automatic Epitope Recognition in Proteins Oriented to the System for Macromolecular Interaction Assessment MIAX Atsushi Yoshimori Carlos A. Del Carpio yosimori@translell.eco.tut.ac.jp
Protein Structures. Sequences of amino acid residues 20 different amino acids. Quaternary. Primary. Tertiary. Secondary. 10/8/2002 Lecture 12 1
 Protein Structures Sequences of amino acid residues 20 different amino acids Primary Secondary Tertiary Quaternary 10/8/2002 Lecture 12 1 Angles φ and ψ in the polypeptide chain 10/8/2002 Lecture 12 2
Protein Structures Sequences of amino acid residues 20 different amino acids Primary Secondary Tertiary Quaternary 10/8/2002 Lecture 12 1 Angles φ and ψ in the polypeptide chain 10/8/2002 Lecture 12 2
Bioinformatics. Scoring Matrices. David Gilbert Bioinformatics Research Centre
 Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
DATE A DAtabase of TIM Barrel Enzymes
 DATE A DAtabase of TIM Barrel Enzymes 2 2.1 Introduction.. 2.2 Objective and salient features of the database 2.2.1 Choice of the dataset.. 2.3 Statistical information on the database.. 2.4 Features....
DATE A DAtabase of TIM Barrel Enzymes 2 2.1 Introduction.. 2.2 Objective and salient features of the database 2.2.1 Choice of the dataset.. 2.3 Statistical information on the database.. 2.4 Features....
Christian Sigrist. November 14 Protein Bioinformatics: Sequence-Structure-Function 2018 Basel
 Christian Sigrist General Definition on Conserved Regions Conserved regions in proteins can be classified into 5 different groups: Domains: specific combination of secondary structures organized into a
Christian Sigrist General Definition on Conserved Regions Conserved regions in proteins can be classified into 5 different groups: Domains: specific combination of secondary structures organized into a
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
QUANTA. Protein Design. Release December Scranton Road San Diego, CA / Fax: 858/
 QUANTA Protein Design Release 2000 December 2000 9685 Scranton Road San Diego, CA 92121-3752 858/799-5000 Fax: 858/799-5100 Copyright * This document is copyright 2001, Accelrys Incorporated. All rights
QUANTA Protein Design Release 2000 December 2000 9685 Scranton Road San Diego, CA 92121-3752 858/799-5000 Fax: 858/799-5100 Copyright * This document is copyright 2001, Accelrys Incorporated. All rights
Procheck output. Bond angles (Procheck) Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.
 Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.") Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Molecular Modeling. Prediction of Protein 3D Structure from Sequence. Vimalkumar Velayudhan. May 21, 2007
 Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm. Alignment scoring schemes and theory: substitution matrices and gap models
 Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm Alignment scoring schemes and theory: substitution matrices and gap models 1 Local sequence alignments Local sequence alignments are necessary
Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm Alignment scoring schemes and theory: substitution matrices and gap models 1 Local sequence alignments Local sequence alignments are necessary
Detection of Protein Binding Sites II
 Detection of Protein Binding Sites II Goal: Given a protein structure, predict where a ligand might bind Thomas Funkhouser Princeton University CS597A, Fall 2007 1hld Geometric, chemical, evolutionary
Detection of Protein Binding Sites II Goal: Given a protein structure, predict where a ligand might bind Thomas Funkhouser Princeton University CS597A, Fall 2007 1hld Geometric, chemical, evolutionary
Sequence comparison: Score matrices
 Sequence comparison: Score matrices http://facultywashingtonedu/jht/gs559_2013/ Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best
Sequence comparison: Score matrices http://facultywashingtonedu/jht/gs559_2013/ Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best
Data analysis and Geostatistics - lecture VI
 Data analysis and Geostatistics - lecture VI Statistical testing with population distributions Statistical testing - the steps 1. Define a hypothesis to test in statistics only a hypothesis rejection is
Data analysis and Geostatistics - lecture VI Statistical testing with population distributions Statistical testing - the steps 1. Define a hypothesis to test in statistics only a hypothesis rejection is
Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent
 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent") Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent concentrations of PcTS (100 µm, blue; 500 µm, green; 1.5 mm,
Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent concentrations of PcTS (100 µm, blue; 500 µm, green; 1.5 mm,
Prediction and Classif ication of Human G-protein Coupled Receptors Based on Support Vector Machines
 Article Prediction and Classif ication of Human G-protein Coupled Receptors Based on Support Vector Machines Yun-Fei Wang, Huan Chen, and Yan-Hong Zhou* Hubei Bioinformatics and Molecular Imaging Key Laboratory,
Article Prediction and Classif ication of Human G-protein Coupled Receptors Based on Support Vector Machines Yun-Fei Wang, Huan Chen, and Yan-Hong Zhou* Hubei Bioinformatics and Molecular Imaging Key Laboratory,
Protein Structure Determination
 Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Resonance assignments in proteins. Christina Redfield
 Resonance assignments in proteins Christina Redfield 1. Introduction The assignment of resonances in the complex NMR spectrum of a protein is the first step in any study of protein structure, function
Resonance assignments in proteins Christina Redfield 1. Introduction The assignment of resonances in the complex NMR spectrum of a protein is the first step in any study of protein structure, function
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
C h a p t e r 2 A n a l y s i s o f s o m e S e q u e n c e... methods use different attributes related to mis sense mutations such as
 C h a p t e r 2 A n a l y s i s o f s o m e S e q u e n c e... 2.1Introduction smentionedinchapter1,severalmethodsareavailabletoclassifyhuman missensemutationsintoeitherbenignorpathogeniccategoriesandthese
C h a p t e r 2 A n a l y s i s o f s o m e S e q u e n c e... 2.1Introduction smentionedinchapter1,severalmethodsareavailabletoclassifyhuman missensemutationsintoeitherbenignorpathogeniccategoriesandthese
Better Bond Angles in the Protein Data Bank
 Better Bond Angles in the Protein Data Bank C.J. Robinson and D.B. Skillicorn School of Computing Queen s University {robinson,skill}@cs.queensu.ca Abstract The Protein Data Bank (PDB) contains, at least
Better Bond Angles in the Protein Data Bank C.J. Robinson and D.B. Skillicorn School of Computing Queen s University {robinson,skill}@cs.queensu.ca Abstract The Protein Data Bank (PDB) contains, at least
NMR, X-ray Diffraction, Protein Structure, and RasMol
 NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
CHAPTER 29 HW: AMINO ACIDS + PROTEINS
 CAPTER 29 W: AMI ACIDS + PRTEIS For all problems, consult the table of 20 Amino Acids provided in lecture if an amino acid structure is needed; these will be given on exams. Use natural amino acids (L)
CAPTER 29 W: AMI ACIDS + PRTEIS For all problems, consult the table of 20 Amino Acids provided in lecture if an amino acid structure is needed; these will be given on exams. Use natural amino acids (L)
Sequence comparison: Score matrices. Genome 559: Introduction to Statistical and Computational Genomics Prof. James H. Thomas
 Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best alignment path onsider the last step in
Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best alignment path onsider the last step in
PAM-1 Matrix 10,000. From: Ala Arg Asn Asp Cys Gln Glu To:
 119-1 atrix 10,000 rom: la rg sn sp ys ln lu o: la 9867 2 9 10 3 8 17 rg 1 9913 1 0 1 10 0 sn 4 1 9822 36 0 4 6 sp 6 0 42 9859 0 6 53 ys 1 1 0 0 9973 0 0 ln 3 9 4 5 0 9876 27 lu 10 0 7 56 0 35 9865 120
119-1 atrix 10,000 rom: la rg sn sp ys ln lu o: la 9867 2 9 10 3 8 17 rg 1 9913 1 0 1 10 0 sn 4 1 9822 36 0 4 6 sp 6 0 42 9859 0 6 53 ys 1 1 0 0 9973 0 0 ln 3 9 4 5 0 9876 27 lu 10 0 7 56 0 35 9865 120
Sequence Bioinformatics. Multiple Sequence Alignment Waqas Nasir
 Sequence Bioinformatics Multiple Sequence Alignment Waqas Nasir 2010-11-12 Multiple Sequence Alignment One amino acid plays coy; a pair of homologous sequences whisper; many aligned sequences shout out
Sequence Bioinformatics Multiple Sequence Alignment Waqas Nasir 2010-11-12 Multiple Sequence Alignment One amino acid plays coy; a pair of homologous sequences whisper; many aligned sequences shout out
Supplementary Figure 1 Crystal contacts in COP apo structure (PDB code 3S0R)
") Supplementary Figure 1 Crystal contacts in COP apo structure (PDB code 3S0R) Shown in cyan and green are two adjacent tetramers from the crystallographic lattice of COP, forming the only unique inter-tetramer
Supplementary Figure 1 Crystal contacts in COP apo structure (PDB code 3S0R) Shown in cyan and green are two adjacent tetramers from the crystallographic lattice of COP, forming the only unique inter-tetramer
Prediction of the subcellular location of apoptosis proteins based on approximate entropy
 Prediction of the subcellular location of apoptosis proteins based on approximate entropy Chaohong Song Feng Shi* Corresponding author Xuan Ma College of Science, Huazhong Agricultural University, Wuhan,
Prediction of the subcellular location of apoptosis proteins based on approximate entropy Chaohong Song Feng Shi* Corresponding author Xuan Ma College of Science, Huazhong Agricultural University, Wuhan,
mechanisms: Discrete molecular dynamics study
 Elucidation of amyloid β -protein oligomerization mechanisms: Discrete molecular dynamics study B. Urbanc 1, M. Betnel 2, L. Cruz 1, G. Bitan 3,4, and D. B. Teplow 3,4 1 Department of Physics, Drexel University,
Elucidation of amyloid β -protein oligomerization mechanisms: Discrete molecular dynamics study B. Urbanc 1, M. Betnel 2, L. Cruz 1, G. Bitan 3,4, and D. B. Teplow 3,4 1 Department of Physics, Drexel University,
Interfacing chemical biology with the -omic sciences and systems biology
 Volume 12 Number 3 March 2016 Pages 681 1058 Molecular BioSystems Interfacing chemical biology with the -omic sciences and systems biology www.rsc.org/molecularbiosystems ISSN 1742-206X PAPER A. Keith
Volume 12 Number 3 March 2016 Pages 681 1058 Molecular BioSystems Interfacing chemical biology with the -omic sciences and systems biology www.rsc.org/molecularbiosystems ISSN 1742-206X PAPER A. Keith
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
BLAST: Target frequencies and information content Dannie Durand
 Computational Genomics and Molecular Biology, Fall 2016 1 BLAST: Target frequencies and information content Dannie Durand BLAST has two components: a fast heuristic for searching for similar sequences
Computational Genomics and Molecular Biology, Fall 2016 1 BLAST: Target frequencies and information content Dannie Durand BLAST has two components: a fast heuristic for searching for similar sequences
Viewing and Analyzing Proteins, Ligands and their Complexes 2
 2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation
 Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
Supplemental Information for. Quaternary dynamics of B crystallin as a direct consequence of localised tertiary fluctuations in the C terminus
 Supplemental Information for Quaternary dynamics of B crystallin as a direct consequence of localised tertiary fluctuations in the C terminus Andrew J. Baldwin 1, Gillian R. Hilton 2, Hadi Lioe 2, Claire
Supplemental Information for Quaternary dynamics of B crystallin as a direct consequence of localised tertiary fluctuations in the C terminus Andrew J. Baldwin 1, Gillian R. Hilton 2, Hadi Lioe 2, Claire
Sunhats for plants. How plants detect dangerous ultraviolet rays
 Sunhats for plants How plants detect dangerous ultraviolet rays Anyone who has ever suffered sunburn will know about the effects of too much ultraviolet (UV) radiation, in particular UV-B (from 280-315
Sunhats for plants How plants detect dangerous ultraviolet rays Anyone who has ever suffered sunburn will know about the effects of too much ultraviolet (UV) radiation, in particular UV-B (from 280-315
Protein Structure Prediction Using Neural Networks
 Protein Structure Prediction Using Neural Networks Martha Mercaldi Kasia Wilamowska Literature Review December 16, 2003 The Protein Folding Problem Evolution of Neural Networks Neural networks originally
Protein Structure Prediction Using Neural Networks Martha Mercaldi Kasia Wilamowska Literature Review December 16, 2003 The Protein Folding Problem Evolution of Neural Networks Neural networks originally
Molecular Modeling Lecture 11 side chain modeling rotamers rotamer explorer buried cavities.
 Molecular Modeling 218 Lecture 11 side chain modeling rotamers rotamer explorer buried cavities. Sidechain Rotamers Discrete approximation of the continuous space of backbone angles. Sidechain conformations
Molecular Modeling 218 Lecture 11 side chain modeling rotamers rotamer explorer buried cavities. Sidechain Rotamers Discrete approximation of the continuous space of backbone angles. Sidechain conformations
SUPPLEMENTARY INFORMATION
 In the format provided by the authors and unedited. SUPPLEMENTARY INFORMATION DOI: 10.1038/NCHEM.2720 1 2 3 Tuning underwater adhesion with cation-π interactions Matthew A. Gebbie, Wei Wei, Alex M. Schrader,
In the format provided by the authors and unedited. SUPPLEMENTARY INFORMATION DOI: 10.1038/NCHEM.2720 1 2 3 Tuning underwater adhesion with cation-π interactions Matthew A. Gebbie, Wei Wei, Alex M. Schrader,
Ch. 9 Multiple Sequence Alignment (MSA)
") Ch. 9 Multiple Sequence Alignment (MSA) - gather seqs. to make MSA - doing MSA with ClustalW - doing MSA with Tcoffee - comparing seqs. that cannot align Introduction - from pairwise alignment to MSA -
Ch. 9 Multiple Sequence Alignment (MSA) - gather seqs. to make MSA - doing MSA with ClustalW - doing MSA with Tcoffee - comparing seqs. that cannot align Introduction - from pairwise alignment to MSA -
Problem Set 1
 2006 7.012 Problem Set 1 Due before 5 PM on FRIDAY, September 15, 2006. Turn answers in to the box outside of 68-120. PLEASE WRITE YOUR ANSWERS ON THIS PRINTOUT. 1. For each of the following parts, pick
2006 7.012 Problem Set 1 Due before 5 PM on FRIDAY, September 15, 2006. Turn answers in to the box outside of 68-120. PLEASE WRITE YOUR ANSWERS ON THIS PRINTOUT. 1. For each of the following parts, pick
Table S1. Primers used for the constructions of recombinant GAL1 and λ5 mutants. GAL1-E74A ccgagcagcgggcggctgtctttcc ggaaagacagccgcccgctgctcgg
 SUPPLEMENTAL DATA Table S1. Primers used for the constructions of recombinant GAL1 and λ5 mutants Sense primer (5 to 3 ) Anti-sense primer (5 to 3 ) GAL1 mutants GAL1-E74A ccgagcagcgggcggctgtctttcc ggaaagacagccgcccgctgctcgg
SUPPLEMENTAL DATA Table S1. Primers used for the constructions of recombinant GAL1 and λ5 mutants Sense primer (5 to 3 ) Anti-sense primer (5 to 3 ) GAL1 mutants GAL1-E74A ccgagcagcgggcggctgtctttcc ggaaagacagccgcccgctgctcgg
arxiv: v1 [cond-mat.soft] 22 Oct 2007
![arxiv: v1 [cond-mat.soft] 22 Oct 2007](/thumbs/78/78401876.jpg "arxiv: v1 [cond-mat.soft] 22 Oct 2007") Conformational Transitions of Heteropolymers arxiv:0710.4095v1 [cond-mat.soft] 22 Oct 2007 Michael Bachmann and Wolfhard Janke Institut für Theoretische Physik, Universität Leipzig, Augustusplatz 10/11,
Conformational Transitions of Heteropolymers arxiv:0710.4095v1 [cond-mat.soft] 22 Oct 2007 Michael Bachmann and Wolfhard Janke Institut für Theoretische Physik, Universität Leipzig, Augustusplatz 10/11,
Francisco M. Couto Mário J. Silva Pedro Coutinho
 Francisco M. Couto Mário J. Silva Pedro Coutinho DI FCUL TR 03 29 Departamento de Informática Faculdade de Ciências da Universidade de Lisboa Campo Grande, 1749 016 Lisboa Portugal Technical reports are
Francisco M. Couto Mário J. Silva Pedro Coutinho DI FCUL TR 03 29 Departamento de Informática Faculdade de Ciências da Universidade de Lisboa Campo Grande, 1749 016 Lisboa Portugal Technical reports are
Protein Structure Prediction, Engineering & Design CHEM 430
 Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Performing a Pharmacophore Search using CSD-CrossMiner
 Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Aqueous solutions. Solubility of different compounds in water
 Aqueous solutions Solubility of different compounds in water The dissolution of molecules into water (in any solvent actually) causes a volume change of the solution; the size of this volume change is
Aqueous solutions Solubility of different compounds in water The dissolution of molecules into water (in any solvent actually) causes a volume change of the solution; the size of this volume change is