Supporting Information. Copyright Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006
|
|
|
- Preston Wood
- 5 years ago
- Views:
Transcription

1 Supporting Information Copyright Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006
2 A Highly Practical RCM Approach towards a Molecular Building Kit of Spirocyclic Reverse Turn Mimics Holger Bittermann, Frank Böckler, Jürgen Einsiedel and Peter Gmeiner* Department of Medicinal Chemistry, Friedrich Alexander University Erlangen-Nürnberg, Schuhstr. 19 D Erlangen (Germany) Contents: 2D NMR spectra of compound 12 including NOESY correlations (S2-3) Detailed description of the theoretical investigations based on quantum chemical calculations (S4-11) Additional references (S12) S1
3 Fig. S1: COSY and HMQC spectra of 12. S2



4 Fig. S2: NOESY spectrum of 12. Fig. S3: Depiction of diagnostic NOESY signals in 12. S3
5 Table S1: Final Geometries of Spiro-β-lactam, Spiro-γ-lactam and Spiro-dehydroloactams after DFT Optimizations at the B3LYP/6-311G(d,p) Level of Theory Revealing Differences in Structural Key-Parameters: Structure spiro-βlactam (3a) spiro-γlactam (3b) [4.5]-spirodehydrolactam (11) [4.6]-spirodehydrolactam (12) [4.7]-spirodehydrolactam (13) Conformation boat 1 boat 2 Ψ i+1 (N i+1, C α i+1, C i+1, N i+2 ) β (C i, C α i+1, C α i+2, N i+3 ) D (O i H i+3 ) Å Å Å Å Å Å D (O i N i+3 ) Å Å Å Å Å Å α (C i =O i H i+3 ) α (O i H i+3 N i+3 ) Structure [4.7]-spirodehydrolactam (13) [4.8]-spiro-dehydrolactam (14) Conformation chair 1 chair 2 conf 1 conf 2 Ψ i+1 (N i+1, C α i+1, C i+1, N i+2 ) β (C i, C α i+1, C α i+2, N i+3 ) β- turn γ- turn D (O i H i+3 ) Å Å Å Å Å D (O i N i+3 ) Å Å Å Å Å α (C i =O i H i+3 ) α (O i H i+3 N i+3 ) S4
6 Detailed description of the theoretical investigations based on quantum chemical calculations: The structure of the spiro-β-lactam (3a) was derived from X-ray data of a suitable precursor, which was further used as a template to build the spiro-γ-lactam (3b) and the highermembered rings of the spiro-dehydrolactam series (11-14) from. For the 7-membered spirodehydrolactam (12), available NOESY information was applied as a filter rule in an initial low level conformational sampling. Thereby, one class of conformations was easily identifiable to yield the best representation of the experimental data. For the 8-membered spirodehydrolactam (13), five discrete confomers were retrieved from initial conformational sampling and all of them were decided to be pursued to facilitate the process of structural determination. Two of these conformers had a boat-like, two a chair-like and one a twisted structure in the 8-membered ring. The twisted structure has turned out to be not a stable discrete minimum in subsequent calculations and, thus, showed a smooth transition into the boat2-conformer. Consequently, only the four persistent conformers are described in TableS1. In the case of the 9-membered spiro-dehydrolactam (14), heterogeneity of structures was anticipated from the spectroscopic data. Thus, the two predominantely occuring conformations were selected from preliminary conformational sampling to be the most plausible structures. In summary, a total of 11 structures was submitted to a series of DFT calculations using Gaussian98 [S1]. The applied optimization protocol has proven to give reasonable results in several previous studies. [S2,3] After a short semiempirial preoptimization (PM3), we used a B3LYP density functional with a 3-21G basis set in order to produce a reasonable geometry in appropriate time. Then, we increased the basis set in three subsequent steps to enhance the quality of the structure, first to the double-valence d-polarized level 6-31G(d), then to the triple-valence d-polarized level 6-311G(d), and finally to the d,p-polarized level G(d,p) additionally adding a diffuse function. At this level of calculation a difference of 8.5 was found between the Ψ i+1 angle of 3a and 3b, comprising that the spiro-β-lactam (3a) considerably approaches the Ψ i+1 angle (120 ) of an ideal type II β-turn. Likewise, the β angle defined by Ball et al. [S4] as the torsion between C i, C α i+1, C α i+2, and N i+3 is more tending towards the ideal value of 0 for 3a (11.8 ) than for 3b (21.2 ). Interestingly, Ψ i+1 and β for the [4.5]-spirodehydrolactam (11) is found in between those of 3a and 3b, with the 6-membered ring showing a Ψ i+1 of and a β of Increasing the ring size to the [4.6]-spirodehydrolactam (12) yields Ψ i+1 and β values of and 21.2, respectively, which are closely appoaching the values of the five-membered ring system 3b. For the [4.7]-spirodehydrolactam (13) the structural properties are strongly dependent on the conformation of the 8-membered ring. While the boat 1 and chair 1 conformations significantly increase both Ψ i+1 and β, the boat 2 conformation adopts an almost ideal Ψ i+1 of and β of 4.4 and, thus, is second to none in the entire series. The chair 2 conformer even decreases Ψ i+1 and β below 120 and 0, respectively. The conformer 1 of the [4.8]-spirodehydrolactam (14) adopts through its further increased ring size S5
7 very high Ψ i+1 and β values and can be regarded as a logical extension of the boat 1 and chair 1 conformers of 13. However, the increased flexibility in the 9-membered ring of 14 yields with conformation 2 a totally different structure, revealing most consistency with the chair 2 conformation of 13. While in all other structures (3a-b,11-13) very reasonable hydrogen-bonding parameters are found, 14 is the only one for which a minimized state (conf. 1) was identified to form more a γ- than a β-turn. This result indicates that 14 may exist in an equilibrium between a β-turn, a γ-turn and even an open state, a conclusion being also in good correspondance to the experimental findings for this compound. Conversely, this means that 14 has also a high ability to adapt to specific binding requirements when integrated in to a peptide-mimetic ligand for a particular target. For 3a, 3b, and conformer 2 of 14, the hydrogen bond distance ( Å) between O i and H i+3, as well as the distance between O i and N i+3 ( Å), is found to be in the required range for a typical H- bond. Moreover, for these structures, also the bond angle α(o i H i+3 N i+3 ), which is ordinarily restricted to values > 150 for a stable hydrogen bond, is in the narrow range of satisfying also this structural requirement. To clarify the relevance of the calculated conformers of 13 and 14, we subjected the optimized structures to an additional series of single point calculations modifying the density functional to B3PW91, enhancing the basis set with additional polarization functions to G(2d,p) and G(2df,2p), as well as treating electron correlation (alternatively to DFT) with second-order Møller-Plesset theory (MP2). Calculations on all levels are subsumed in Table S2. These calculations were found to give a highly consistent ranking of the conformers with almost identical relative energy differences. For the [4.7]-spirodehydrolactam (13), the boat2 conformation with its planar olefinic part folded away from the hydrogen bond (see Figure S4) is in general the most favoured conformer, followed by boat1 with the planar olefinic part folded towards the hydrogen bond. On all levels except for the MP2 electron correlation, the relative energy difference of boat2 versus boat1 is approximately 3 kcal/mol in favour of boat2, while this difference is reduced to 1.6 kcal/mol for MP2. This indicates that according to the Boltzmann equation, the boat1 conformation could be populated in the range of 0.5% to 7%. However, regarding the high conformational strain of this molecule, the transition state barrier between boat1 and boat2 should be considerably high. As the chair conformations are substantially more unfavourable exhibiting an energy difference to boat2 of about 7 kcal/mol (chair1) or 10 kcal/mol (chair2), we suggest that boat2 should be the predominantly found conformation (> 90%). S6
8 Table S2: Absolute energy [hartree] of the preferential conformer and relative energy differences [kcal/mol] towards the other conformers calculated at various levels of theory: [4.7]-spiro-dehydrolactam (13): method (density functional) / basis set E boat2 [hartree] ΔE boat1-boat2 [kcal/mol] ΔE chair1-boat2 [kcal/mol] ΔE chair2-boat2 [kcal/mol] B3LYP/3-21G Opt B3LYP/6-31G(d) Opt B3LYP/6-311G(d) Opt B3LYP/6-311+G(d,p) Opt B3PW91/6-311+G(2d,p) SP B3PW91/6-311+G(2df,2p) SP MP2/6-311+G(2d,p) SP [4.8]-spiro-dehydrolactam (14): method (density functional) / basis set E conf1 [hartree] ΔE conf2-conf1 [kcal/mol] B3LYP/3-21G Opt B3LYP/6-31G(d) Opt B3LYP/6-311G(d) Opt B3LYP/6-311+G(d,p) Opt B3PW91/6-311+G(2d,p) SP B3PW91/6-311+G(2df,2p) SP MP2/6-311+G(2d,p) SP S7

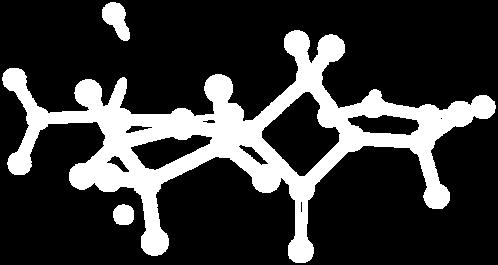
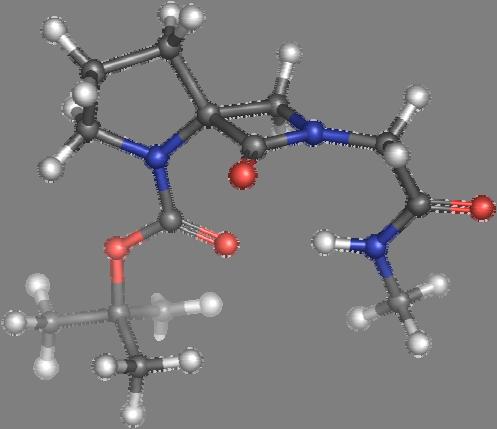
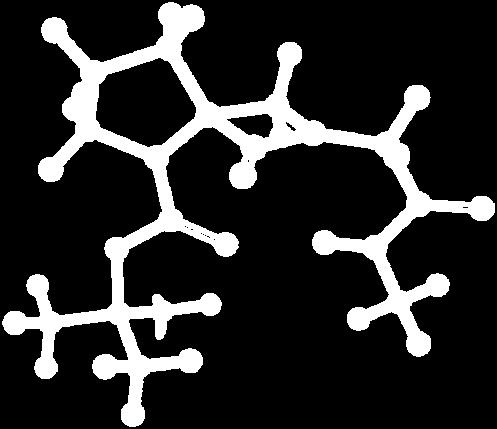
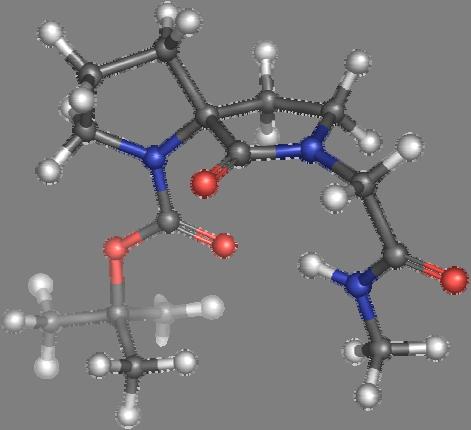
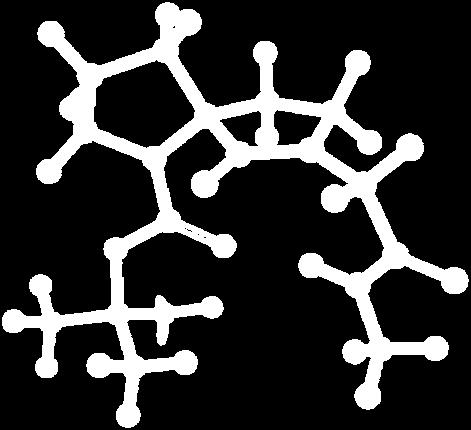
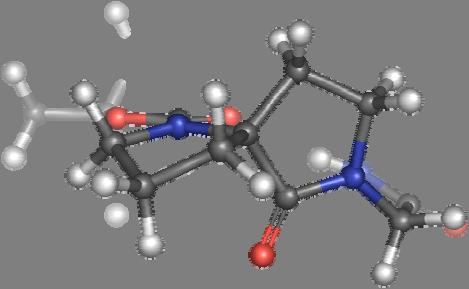
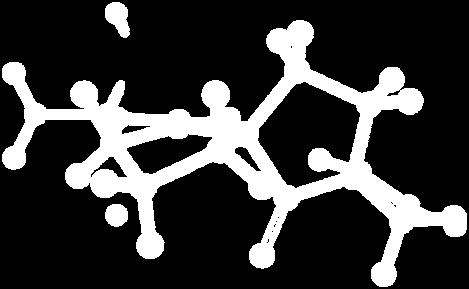
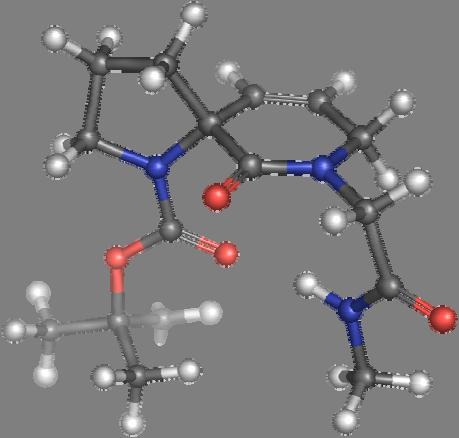
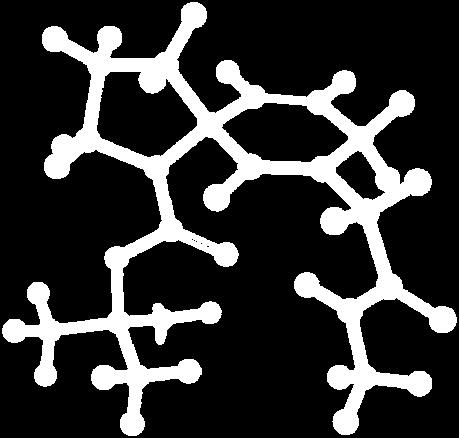
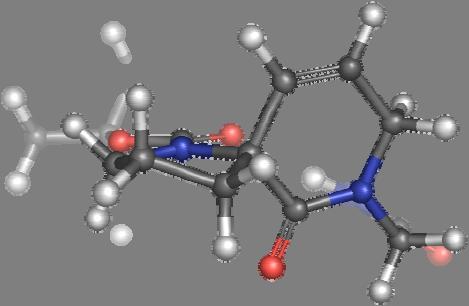
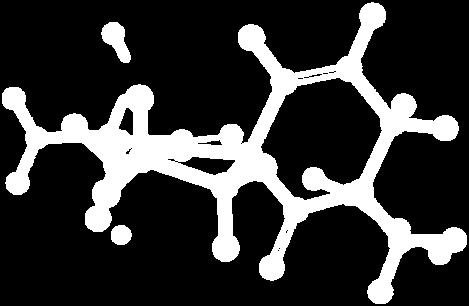
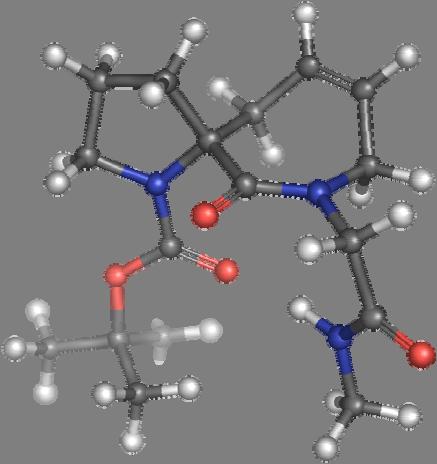
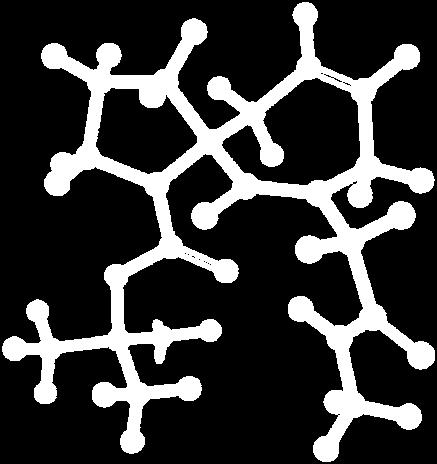
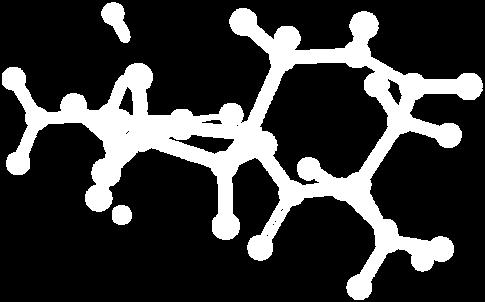
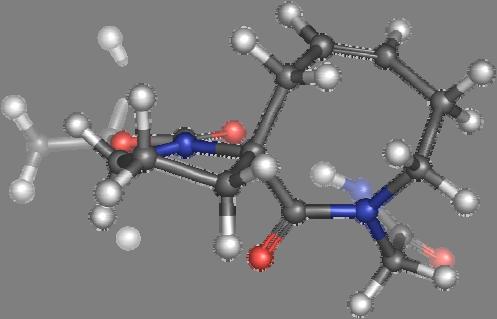
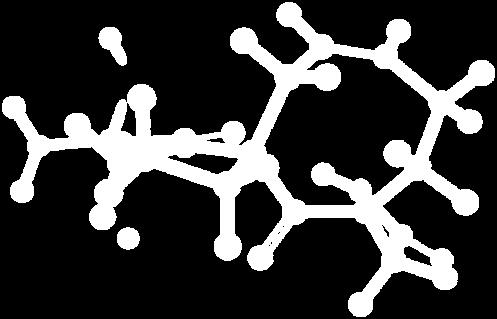
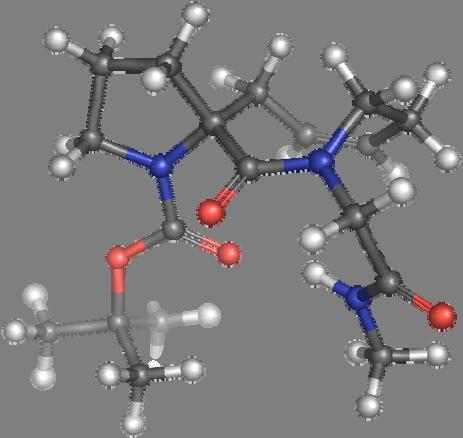
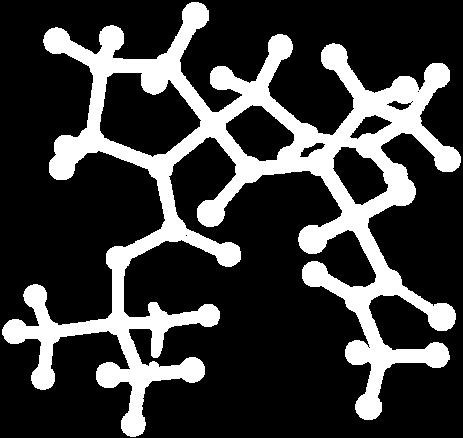
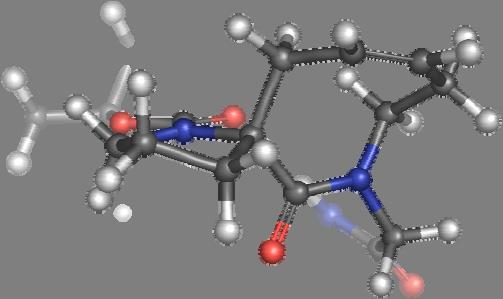

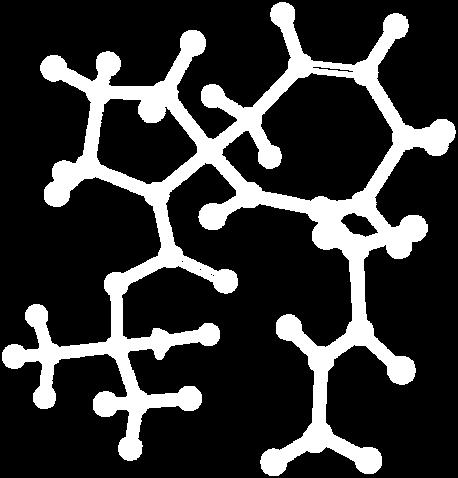
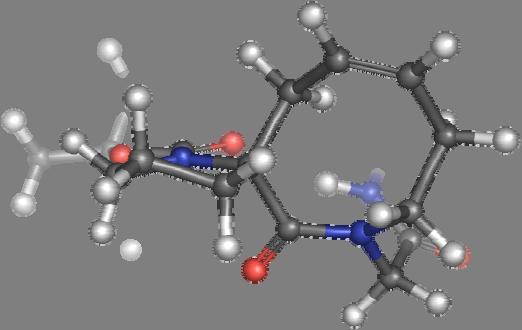
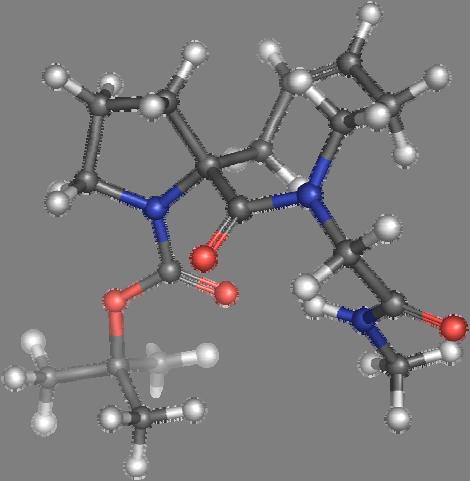
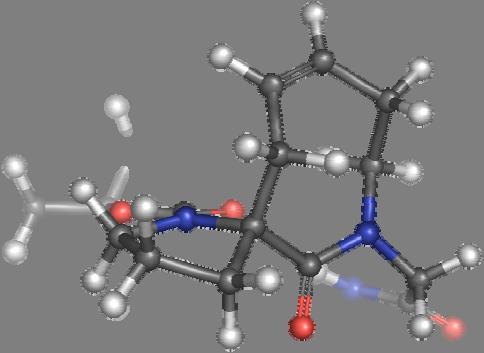
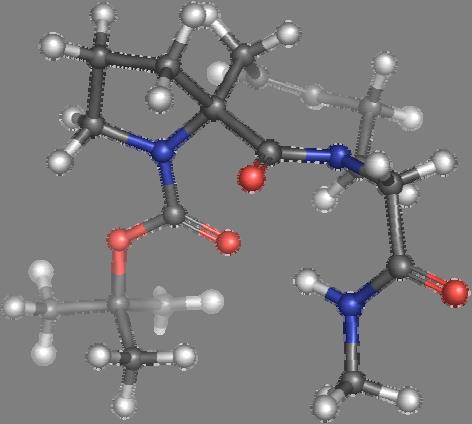
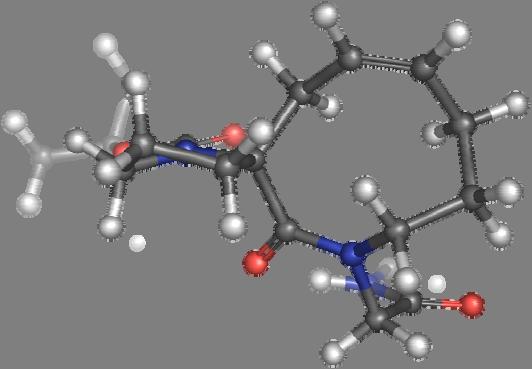
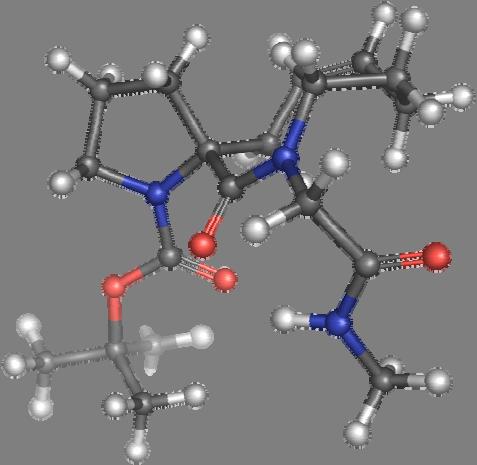
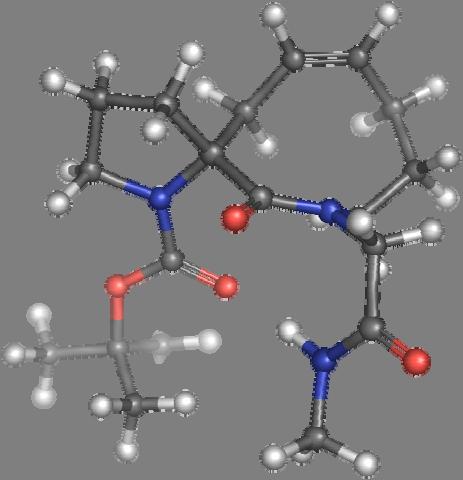
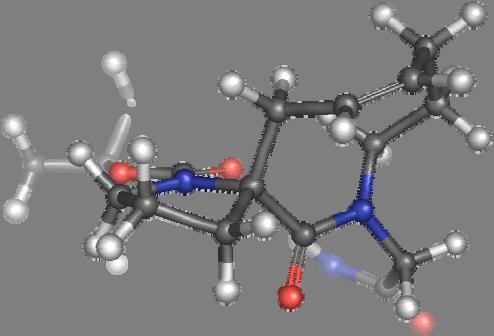

9 Figure S4: Structures 3a,b and optimized by DFT-calculations ( B3LYP/6-311+G(d,p) ): Spiro-β-lactam (3a) boat 1 Spiro-γ-lactam (3b) [4.5]spirodehydrolactam (11) [4.7]spiro-dehydrolactam (13) boat 2 chair 1 [4.8]spiro-dehydrolactam (14) conformer 1 conformer 2 [4.6]spirodehydrolactam (12) chair 2 S8

![Therefore, we calculated the magnetic shielding tensor using gauge invariant atomic orbitals [S5] (GIAO) within B3PW91 / 6-311+G(2d,p) single point calculation for all four conformations.](/docs-images/92/109964154/images/10-1.jpg "As outlined in Table S3+4, considerable differences in absolute and relative chemical shifts were obtained for the four conformers.")
 and, thus, calculated their chemical shifts by subtraction of the total shielding (average isotropic value) of the respective carbon atom from the total shielding of a TMS-carbon atom: δ α α 8 9")

10 In order to facilitate the interpretation of our NMR-spectroscopic investigations and to corroborate the quality of the obtained structures, we have checked the conformity of their calculated magnetic shielding versus the experimental NMR data. Therefore, we calculated the magnetic shielding tensor using gauge invariant atomic orbitals [S5] (GIAO) within B3PW91 / G(2d,p) single point calculation for all four conformations. As outlined in Table S3+4, considerable differences in absolute and relative chemical shifts were obtained for the four conformers. As the structural differences between boat 1, boat 2, chair 1 and chair 2 predominantly influence the hydrogens at the 8-membered ring, we have employed them as state-specific NMR-probes (see Figure S5) and, thus, calculated their chemical shifts by subtraction of the total shielding (average isotropic value) of the respective carbon atom from the total shielding of a TMS-carbon atom: δ α α = σ σ x 8 { } ; C H, C H', C H, C H', C H, C H', C H, C H, C H, C H' TMS x TMS as a reference was optimized and subjected to NMR single point calculations on the same levels as the compared structures utilizing its T d -symmetry. In addition to these absolute chemical shift values, the relative separation (Δδ) of the geminal hydrogen pairs (H and H ) as well as of the pair of olefinic hydrogens was used as another validation criteria, which has proven to be very useful for the comparison with experimental data. [S2,3] Figure S5 Structure of 13 in the boat 2 conformation. The hydrogen atoms used as state-specific NMRprobes are indicated with yellow spheres and labeled according to the numbering in Table S3+4. S9
11 Table S3: Comparison between experimental and calculated absolute chemical shifts δ [ppm] for four conformers of the [4.7]-spirodehydrolactam (13) Hydrogen atoms Exp. δ Predicted abs. shift δ [ppm] δ exp - δ calc [ppm] 298 K 220 K 298 K 220 K bt 1 bt 2 ch 1 ch 2 bt 1 bt 2 ch 1 ch 2 bt 1 bt 2 ch 1 ch 2 C α H C α H C 8 H C 8 H C 9 H C 9 H C 10 H C 11 H C 12 H C 12 H mean Table S4: Comparison between experimental and calculated relative chemical shift differences Δδ [ppm] for four conformers of the [4.7]-spirodehydrolactam (13) Compared hydrogens Exp. Δδ Predicted abs. shift δ [ppm] δ exp - δ calc [ppm] 298 K 220 K 298 K 220 K bt 1 bt 2 ch 1 ch 2 bt 1 bt 2 ch 1 ch 2 bt 1 bt 2 ch 1 ch 2 C α H-C α H C 8 H-C 8 H C 9 H-C 9 H C 12 H-C 12 H mean S10
12 In the comparison of absolute chemical shifts (Table S3) the predicted values of the boat 2 conformer show clearly the best correspondance to the experimental data both determined at 298K (mean deviation: 0.16) and 220K (mean deviation: 0.13). This result becomes even more obvious, when focussing on the hydrogens attached to C 8 only, as their chemical shifts show the strongest dependence on the evaluated conformation. Only for boat 2 an almost quantitative prediction of the chemical shift for C 8 H (deviation: 0.15 ppm at 298 K) and C 8 H (deviation: 0.14 ppm at 298 K) is possible. The deviations of these hydrogens even further decrease to 0.11 ppm for C 8 H and 0.01 ppm for C 8 H, when the NMR measuring conditions are cooled down to 220 K leading to more sharpened signals with a slight shift of δ towards a stronger separation of the signals. Thus, we conclude that at 298 K there is a slow equilibrium between the dominant boat 2 conformer and a higher energy conformer leading to movement in particular in the area of C 8, where the hydrogen signals are broadened. One putative candidate for such a higher energy local minimum could be similar to the twisted structure, which showed a transition into the boat 2 conformer during early stages of DFTminimization. When cooling to 220 K, the signals sharpen and are even better represented by the boat 2 conformer, which is likely to be the only conformation existing at lower temperatures. When looking at the relative separation of the chemical shifts of C 8 H and C 8 H, the dominance of the boat 2 versus the other conformers is clarified even more: While the experimental separation of 2.06 ppm at 220 K is almost exactly predicted by the boat 2 conformer with a deviation of 0.12 ppm, the other conformers do not even come close to this value (0.68 ppm for boat 1, 0.84 ppm for chair 2 and 1.82 ppm for chair 1). In conclusion, based on its global minimum energy, as well as an almost ideal correspondance to experimental 1 H-NMR data, we suggest that boat 2 is the predominant conformer. For the [4.8]-spiro-dehydrolactam (14), the low energy gap found between conformer 1 and 2, which we calculated on the same levels of theory as the conformers of 13 (Table S2), indicates that 14 exists in an equilibrium of at least these two conformers at room temperature. Due to our calculations, conformer 1 tends to be preferred over conformer 2 by about 1.1 to 2.0 kcal/mol, however the increase of the ring size will lower the energy barriers between these (and perhaps other) states. This corresponds to the existence of broadened signals in the experimental data, suggesting a slow exchange between different conformational states. S11
13 Additional References: [S1] Gaussian 98 (Revision A.7), M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. J. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle, J. A. Pople, Gaussian, Inc., Pittsburgh, PA, [S2] C. Lenz, F. Boeckler, H. Hübner, P. Gmeiner, Bioorg Med Chem 2004, 12, 113. [S3] C. Lenz, F. Boeckler, H. Hübner, P. Gmeiner, Bioorg Med Chem 2005, 13, [S4] J. B. Ball, R. A. Hughes, P. F. Alewood, P. R. Andrews, Tetrahedron 1993, 49, [S5] K. Wolinski, J. F. Hinton, P. Pulay, Journal of the American Chemical Society 1990, 112, 825 S12
Ferromagnetic Coupling of [Ni(dmit) 2 ] - Anions in. (m-fluoroanilinium)(dicyclohexano[18]crown-6)[ni(dmit) 2 ]
![Ferromagnetic Coupling of [Ni(dmit) 2 ] - Anions in. (m-fluoroanilinium)(dicyclohexano[18]crown-6)[ni(dmit) 2 ]](/thumbs/93/117991155.jpg "Ferromagnetic Coupling of [Ni(dmit) 2 ] - Anions in. (m-fluoroanilinium)(dicyclohexano[18]crown-6)[ni(dmit) 2 ]") Supporting Information Ferromagnetic Coupling of [Ni(dmit) 2 ] - Anions in (m-fluoroanilinium)(dicyclohexano[18]crown-6)[ni(dmit) 2 ] Tomoyuki Akutagawa, *,, Daisuke Sato, Qiong Ye, Shin-ichiro Noro,,
Supporting Information Ferromagnetic Coupling of [Ni(dmit) 2 ] - Anions in (m-fluoroanilinium)(dicyclohexano[18]crown-6)[ni(dmit) 2 ] Tomoyuki Akutagawa, *,, Daisuke Sato, Qiong Ye, Shin-ichiro Noro,,
Superacid promoted reactions of N-acyliminium salts and evidence for the involvement of superelectrophiles
 Superacid promoted reactions of N-acyliminium salts and evidence for the involvement of superelectrophiles Yiliang Zhang, Daniel J. DeSchepper, Thomas M. Gilbert, and Douglas A. Klumpp Department of Chemistry
Superacid promoted reactions of N-acyliminium salts and evidence for the involvement of superelectrophiles Yiliang Zhang, Daniel J. DeSchepper, Thomas M. Gilbert, and Douglas A. Klumpp Department of Chemistry
Supporting Information
 Supporting Information Hydrogen-bonding Interactions Between [BMIM][BF 4 ] and Acetonitrile Yan-Zhen Zheng, a Nan-Nan Wang, a,b Jun-Jie Luo, a Yu Zhou a and Zhi-Wu Yu*,a a Key Laboratory of Bioorganic
Supporting Information Hydrogen-bonding Interactions Between [BMIM][BF 4 ] and Acetonitrile Yan-Zhen Zheng, a Nan-Nan Wang, a,b Jun-Jie Luo, a Yu Zhou a and Zhi-Wu Yu*,a a Key Laboratory of Bioorganic
Ab Initio and Density Functional Study
 29 Si NMR Chemical Shifts of Siloxanes: Ab Initio and Density Functional Study Georgios Tsantes, Norbert Auner,* Thomas Müller* Institut für Anorganische Chemie, Johann Wolfgang Goethe-Universität Frankfurt
29 Si NMR Chemical Shifts of Siloxanes: Ab Initio and Density Functional Study Georgios Tsantes, Norbert Auner,* Thomas Müller* Institut für Anorganische Chemie, Johann Wolfgang Goethe-Universität Frankfurt
Analysis of Permanent Electric Dipole Moments of Aliphatic Amines.
 Analysis of Permanent Electric Dipole Moments of Aliphatic Amines. Boris Lakard* LPUB, UMR CNRS 5027, University of Bourgogne, F-21078, Dijon, France Internet Electronic Conference of Molecular Design
Analysis of Permanent Electric Dipole Moments of Aliphatic Amines. Boris Lakard* LPUB, UMR CNRS 5027, University of Bourgogne, F-21078, Dijon, France Internet Electronic Conference of Molecular Design
Supplementary information
 Supplementary information doi: 10.1038/nchem.287 A Potential Energy Surface Bifurcation in Terpene Biosynthesis Young J. Hong and Dean J. Tantillo* Department of Chemistry, University of California, Davis,
Supplementary information doi: 10.1038/nchem.287 A Potential Energy Surface Bifurcation in Terpene Biosynthesis Young J. Hong and Dean J. Tantillo* Department of Chemistry, University of California, Davis,
3,4-Ethylenedioxythiophene (EDOT) and 3,4- Ethylenedioxyselenophene (EDOS): Synthesis and Reactivity of
 and 3,4- Ethylenedioxyselenophene (EDOS): Synthesis and Reactivity of") Supporting Information 3,4-Ethylenedioxythiophene (EDOT) and 3,4- Ethylenedioxyselenophene (EDOS): Synthesis and Reactivity of C α -Si Bond Soumyajit Das, Pradip Kumar Dutta, Snigdha Panda, Sanjio S. Zade*
Supporting Information 3,4-Ethylenedioxythiophene (EDOT) and 3,4- Ethylenedioxyselenophene (EDOS): Synthesis and Reactivity of C α -Si Bond Soumyajit Das, Pradip Kumar Dutta, Snigdha Panda, Sanjio S. Zade*
Supporting Information
 Supporting Information Wiley-VCH 2007 69451 Weinheim, Germany From the alkyllithium aggregate [(nbuli) 2 PMDTA] 2 to lithiated PMDTA Carsten Strohmann*, Viktoria H. Gessner Institut für Anorganische Chemie,
Supporting Information Wiley-VCH 2007 69451 Weinheim, Germany From the alkyllithium aggregate [(nbuli) 2 PMDTA] 2 to lithiated PMDTA Carsten Strohmann*, Viktoria H. Gessner Institut für Anorganische Chemie,
Decomposition!of!Malonic!Anhydrides. Charles L. Perrin,* Agnes Flach, and Marlon N. Manalo SUPPORTING INFORMATION
 S1 Decomposition!of!Malonic!Anhydrides Charles L. Perrin,* Agnes Flach, and Marlon N. Manalo SUPPORTING INFORMATION Complete Reference 26: M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M.
S1 Decomposition!of!Malonic!Anhydrides Charles L. Perrin,* Agnes Flach, and Marlon N. Manalo SUPPORTING INFORMATION Complete Reference 26: M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M.
A dominant homolytic O-Cl bond cleavage with low-spin triplet-state Fe(IV)=O formed is revealed in the mechanism of heme-dependent chlorite dismutase
=O formed is revealed in the mechanism of heme-dependent chlorite dismutase") Supplementary Information to: A dominant homolytic O-Cl bond cleavage with low-spin triplet-state Fe(IV)=O formed is revealed in the mechanism of heme-dependent chlorite dismutase Shuo Sun, Ze-Sheng Li,
Supplementary Information to: A dominant homolytic O-Cl bond cleavage with low-spin triplet-state Fe(IV)=O formed is revealed in the mechanism of heme-dependent chlorite dismutase Shuo Sun, Ze-Sheng Li,
Truong Ba Tai, Long Van Duong, Hung Tan Pham, Dang Thi Tuyet Mai and Minh Tho Nguyen*
 Supplementary Information: A Disk-Aromatic Bowl Cluster B 30 : Towards Formation of Boron Buckyballs Truong Ba Tai, Long Van Duong, Hung Tan Pham, Dang Thi Tuyet Mai and Minh Tho Nguyen* The file contains
Supplementary Information: A Disk-Aromatic Bowl Cluster B 30 : Towards Formation of Boron Buckyballs Truong Ba Tai, Long Van Duong, Hung Tan Pham, Dang Thi Tuyet Mai and Minh Tho Nguyen* The file contains
Supporting Information. for. Silylation of Iron-Bound Carbon Monoxide. Affords a Terminal Fe Carbyne
 Supporting Information for Silylation of Iron-Bound Carbon Monoxide Affords a Terminal Fe Carbyne Yunho Lee and Jonas C. Peters* Division of Chemistry and Chemical Engineering, California Institute of
Supporting Information for Silylation of Iron-Bound Carbon Monoxide Affords a Terminal Fe Carbyne Yunho Lee and Jonas C. Peters* Division of Chemistry and Chemical Engineering, California Institute of
Planar Pentacoordinate Carbon in CAl 5 + : A Global Minimum
 Supporting Information: Planar Pentacoordinate Carbon in CAl 5 + : A Global Minimum Yong Pei, Wei An, Keigo Ito, Paul von Ragué Schleyer, Xiao Cheng Zeng * Department of Chemistry and Nebraska Center for
Supporting Information: Planar Pentacoordinate Carbon in CAl 5 + : A Global Minimum Yong Pei, Wei An, Keigo Ito, Paul von Ragué Schleyer, Xiao Cheng Zeng * Department of Chemistry and Nebraska Center for
3D Structure Based Atomic Charge Calculation for Molecular Mechanics and Molecular Dynamics Simulations
 3D Structure Based tomic Charge Calculation for Molecular Mechanics and Molecular Dynamics Simulations Tatsuya Nakano a*, Tsuguchika Kaminuma a, Masami Uebayasi b, and Yoshiro Nakata c a Division of Chem-Bio
3D Structure Based tomic Charge Calculation for Molecular Mechanics and Molecular Dynamics Simulations Tatsuya Nakano a*, Tsuguchika Kaminuma a, Masami Uebayasi b, and Yoshiro Nakata c a Division of Chem-Bio
Methionine Ligand selectively promotes monofunctional adducts between Trans-EE platinum anticancer drug and Guanine DNA base
 Supplementary Material (ESI) for Chemical Communications This journal is The Royal Society of Chemistry 2010 Supplementary Information Methionine Ligand selectively promotes monofunctional adducts between
Supplementary Material (ESI) for Chemical Communications This journal is The Royal Society of Chemistry 2010 Supplementary Information Methionine Ligand selectively promotes monofunctional adducts between
Supporting Information
 Supporting Information Wiley-VCH 2007 69451 Weinheim, Germany Aluminum Siting in Silicon-rich Zeolite Frameworks. A Combined High Resolution 27 Al NMR and QM/MM Study of ZSM-5 Stepan Sklenak,* Jiří Dědeček,
Supporting Information Wiley-VCH 2007 69451 Weinheim, Germany Aluminum Siting in Silicon-rich Zeolite Frameworks. A Combined High Resolution 27 Al NMR and QM/MM Study of ZSM-5 Stepan Sklenak,* Jiří Dědeček,
Supplemental Material
 Supplemental Material Sensitivity of Hydrogen Bonds of DNA and RNA to Hydration, as Gauged by 1 JNH Measurements in Ethanol Water Mixtures Marlon N. Manalo, Xiangming Kong, and Andy LiWang* Texas A&M University
Supplemental Material Sensitivity of Hydrogen Bonds of DNA and RNA to Hydration, as Gauged by 1 JNH Measurements in Ethanol Water Mixtures Marlon N. Manalo, Xiangming Kong, and Andy LiWang* Texas A&M University
Ab Initio Molecular Orbital Study of the Reactivity of Active Alkyl Groups. V. Nitrosation Mechanism of Acetone with syn-form of Methyl Nitrite
 1502 Notes Chem. Pharm. Bull. 50(11) 1502 1506 (2002) Vol. 50, No. 11 Ab Initio Molecular Orbital Study of the Reactivity of Active Alkyl Groups. V. Nitrosation Mechanism of Acetone with syn-form of Methyl
1502 Notes Chem. Pharm. Bull. 50(11) 1502 1506 (2002) Vol. 50, No. 11 Ab Initio Molecular Orbital Study of the Reactivity of Active Alkyl Groups. V. Nitrosation Mechanism of Acetone with syn-form of Methyl
Aluminum Siting in the ZSM-5 Framework by Combination of
 Supplementary Information Aluminum Siting in the ZSM-5 Framework by Combination of High Resolution 27 Al NMR and DFT/MM calculations Stepan Sklenak,* a Jiří Dědeček, a Chengbin Li, a Blanka Wichterlová,
Supplementary Information Aluminum Siting in the ZSM-5 Framework by Combination of High Resolution 27 Al NMR and DFT/MM calculations Stepan Sklenak,* a Jiří Dědeček, a Chengbin Li, a Blanka Wichterlová,
Synergistic Effects of Water and SO 2 on Degradation of MIL-125 in the Presence of Acid Gases
 Supporting Information Synergistic Effects of Water and SO 2 on Degradation of MIL-125 in the Presence of Acid Gases William P. Mounfield, III, Chu Han,, Simon H. Pang, Uma Tumuluri, Yang Jiao, Souryadeep
Supporting Information Synergistic Effects of Water and SO 2 on Degradation of MIL-125 in the Presence of Acid Gases William P. Mounfield, III, Chu Han,, Simon H. Pang, Uma Tumuluri, Yang Jiao, Souryadeep
Supporting Information
 Supporting Information Wiley-VCH 2007 69451 Weinheim, Germany The Donor-Acceptor Cyclopropanes as Three-Carbon Component in [4+3]-Cycloaddition. Reaction with 1,3-Diphenylisobenzofuran lga A. Ivanova,*
Supporting Information Wiley-VCH 2007 69451 Weinheim, Germany The Donor-Acceptor Cyclopropanes as Three-Carbon Component in [4+3]-Cycloaddition. Reaction with 1,3-Diphenylisobenzofuran lga A. Ivanova,*
Electronic Supplementary information
 Electronic Supplementary information SERS observation of soft C H vibrational mode of bifunctional alkanethiol molecules adsorbed at Au and Ag electrodes Inga Razmute-Razmė, Zenonas Kuodis, Olegas Eicher-Lorka
Electronic Supplementary information SERS observation of soft C H vibrational mode of bifunctional alkanethiol molecules adsorbed at Au and Ag electrodes Inga Razmute-Razmė, Zenonas Kuodis, Olegas Eicher-Lorka
SUPPORTING INFORMATION
 SUPPORTING INFORMATION Highly Luminescent Tetradentate Bis-Cyclometalated Platinum Complexes: Design, Synthesis, Structure, Photophysics, and Electroluminescence Application Dileep A. K. Vezzu, Joseph
SUPPORTING INFORMATION Highly Luminescent Tetradentate Bis-Cyclometalated Platinum Complexes: Design, Synthesis, Structure, Photophysics, and Electroluminescence Application Dileep A. K. Vezzu, Joseph
Supporting Information
 Electronic Supplementary Material (ESI) for Energy & Environmental Science. This journal is The Royal Society of Chemistry 2014 Supporting Information Perylene Diimides: a Thickness-Insensitive Cathode
Electronic Supplementary Material (ESI) for Energy & Environmental Science. This journal is The Royal Society of Chemistry 2014 Supporting Information Perylene Diimides: a Thickness-Insensitive Cathode
Supporting Information. for. Angew. Chem. Int. Ed. Z Wiley-VCH 2003
 Supporting Information for Angew. Chem. Int. Ed. Z52177 Wiley-VCH 2003 69451 Weinheim, Germany A pair of remarkably stable mononuclear chromium(iii) and chromium(iv) hydrides Alexander C. Filippou,* Sven
Supporting Information for Angew. Chem. Int. Ed. Z52177 Wiley-VCH 2003 69451 Weinheim, Germany A pair of remarkably stable mononuclear chromium(iii) and chromium(iv) hydrides Alexander C. Filippou,* Sven
Spin contamination as a major problem in the calculation of spin-spin coupling in triplet biradicals
 Supporting Information to the manuscript Spin contamination as a major problem in the calculation of spin-spin coupling in triplet biradicals P. Jost and C. van Wüllen Contents Computational Details...
Supporting Information to the manuscript Spin contamination as a major problem in the calculation of spin-spin coupling in triplet biradicals P. Jost and C. van Wüllen Contents Computational Details...
Comparison of molecular structure of alkali metal ortho substituted benzoates
 Spectroscopy 24 (2010) 439 443 439 DOI 10.3233/SPE-2010-0444 IOS Press Comparison of molecular structure of alkali metal ortho substituted benzoates R. Świsłocka Department of Chemistry, Biatystok Technical
Spectroscopy 24 (2010) 439 443 439 DOI 10.3233/SPE-2010-0444 IOS Press Comparison of molecular structure of alkali metal ortho substituted benzoates R. Świsłocka Department of Chemistry, Biatystok Technical
Supporting Information
 Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2015 Supporting Information Prenylated Benzoylphloroglucinols and from the Leaves of Garcinia multiflora
Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2015 Supporting Information Prenylated Benzoylphloroglucinols and from the Leaves of Garcinia multiflora
Electronic supplementary information (ESI) Infrared spectroscopy of nucleotides in the gas phase 2. The protonated cyclic 3,5 -adenosine monophosphate
 Infrared spectroscopy of nucleotides in the gas phase 2. The protonated cyclic 3,5 -adenosine monophosphate") Electronic supplementary information (ESI) Infrared spectroscopy of nucleotides in the gas phase 2. The protonated cyclic 3,5 -adenosine monophosphate Francesco Lanucara, a,b Maria Elisa Crestoni,* a Barbara
Electronic supplementary information (ESI) Infrared spectroscopy of nucleotides in the gas phase 2. The protonated cyclic 3,5 -adenosine monophosphate Francesco Lanucara, a,b Maria Elisa Crestoni,* a Barbara
Supporting Information
 Supporting Information Wiley-VCH 2007 69451 Weinheim, Germany Crossover Site-Selectivity in the Adsorption of the Fullerene Derivative PCBM on Au(111) David Écija, a Roberto Otero, a Luis Sánchez, b José
Supporting Information Wiley-VCH 2007 69451 Weinheim, Germany Crossover Site-Selectivity in the Adsorption of the Fullerene Derivative PCBM on Au(111) David Écija, a Roberto Otero, a Luis Sánchez, b José
Metal Enhanced Interactions of Graphene with Monosaccharides. A Manuscript Submitted for publication to. Chemical Physics Letters.
 Metal Enhanced Interactions of Graphene with Monosaccharides A Manuscript Submitted for publication to Chemical Physics Letters February 15, 2016 Carlos Pereyda-Pierre a and Abraham F. Jalbout b* a DIFUS,
Metal Enhanced Interactions of Graphene with Monosaccharides A Manuscript Submitted for publication to Chemical Physics Letters February 15, 2016 Carlos Pereyda-Pierre a and Abraham F. Jalbout b* a DIFUS,
Calculating Accurate Proton Chemical Shifts of Organic Molecules with Density Functional Methods and Modest Basis Sets
 Calculating Accurate Proton Chemical hifts of rganic Molecules with Density Functional Methods and Modest Basis ets Rupal Jain,, # Thomas Bally,, * and Paul Rablen $, * Department of Chemistry, University
Calculating Accurate Proton Chemical hifts of rganic Molecules with Density Functional Methods and Modest Basis ets Rupal Jain,, # Thomas Bally,, * and Paul Rablen $, * Department of Chemistry, University
Effect of Ionic Size on Solvate Stability of Glyme- Based Solvate Ionic Liquids
 Supporting Information for: Effect of Ionic Size on Solvate Stability of Glyme- Based Solvate Ionic Liquids Toshihiko Mandai,,ǁ Kazuki Yoshida, Seiji Tsuzuki, Risa Nozawa, Hyuma Masu, Kazuhide Ueno, Kaoru
Supporting Information for: Effect of Ionic Size on Solvate Stability of Glyme- Based Solvate Ionic Liquids Toshihiko Mandai,,ǁ Kazuki Yoshida, Seiji Tsuzuki, Risa Nozawa, Hyuma Masu, Kazuhide Ueno, Kaoru
A Complete 1 H and 13 C NMR data assignment for N-Benzo[1,3]dioxol-5-ylmethyl-2-(2,2,2-trichloroacetylamino) benzamide*
![A Complete 1 H and 13 C NMR data assignment for N-Benzo[1,3]dioxol-5-ylmethyl-2-(2,2,2-trichloroacetylamino) benzamide*](/thumbs/85/92077222.jpg "A Complete 1 H and 13 C NMR data assignment for N-Benzo[1,3]dioxol-5-ylmethyl-2-(2,2,2-trichloroacetylamino) benzamide*") A Complete 1 H and 13 C NMR data assignment for N-Benzo[1,3]dioxol... 273 Asian Chemistry Letters Vol. 14, No. 3 (2010) 273-278 A Complete 1 H and 13 C NMR data assignment for N-Benzo[1,3]dioxol-5-ylmethyl-2-(2,2,2-trichloroacetylamino)
A Complete 1 H and 13 C NMR data assignment for N-Benzo[1,3]dioxol... 273 Asian Chemistry Letters Vol. 14, No. 3 (2010) 273-278 A Complete 1 H and 13 C NMR data assignment for N-Benzo[1,3]dioxol-5-ylmethyl-2-(2,2,2-trichloroacetylamino)
Supporting Information. spectroscopy and ab initio calculations of a large. amplitude intramolecular motion
 Supporting Information Pseudorotation in pyrrolidine: rotational coherence spectroscopy and ab initio calculations of a large amplitude intramolecular motion Maksim Kunitski, Christoph Riehn, Victor V.
Supporting Information Pseudorotation in pyrrolidine: rotational coherence spectroscopy and ab initio calculations of a large amplitude intramolecular motion Maksim Kunitski, Christoph Riehn, Victor V.
Effects of Intramolecular Basis Set Superposition Error on Conformational Energy Difference of 1,2-Difluoroethane and 1,2-Dimethoxyethane
 Intramolecular Basis Set Superposition Error Bull. Korean Chem. Soc. 2002, Vol. 23, No. 9 1267 Effects of Intramolecular Basis Set Superposition Error on al Energy Difference of 1,2-Difluoroethane and
Intramolecular Basis Set Superposition Error Bull. Korean Chem. Soc. 2002, Vol. 23, No. 9 1267 Effects of Intramolecular Basis Set Superposition Error on al Energy Difference of 1,2-Difluoroethane and
Supplementary Material
 This journal is the wner Societies 00 Macholl et al., Trityl biradicals and C Dynamic uclear Polarization, PCCP 00 Supplementary Material. -dimensional structure of the trityl monoradical c b a d Gaussian98
This journal is the wner Societies 00 Macholl et al., Trityl biradicals and C Dynamic uclear Polarization, PCCP 00 Supplementary Material. -dimensional structure of the trityl monoradical c b a d Gaussian98
Supporting Information. Copyright Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008
 Supporting Information Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2008 Pyridine Catalyzed Stereoselective Addition of Acyclic 1,2-Diones to Acetylenic Ester: Synthetic and Theoretical
Supporting Information Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2008 Pyridine Catalyzed Stereoselective Addition of Acyclic 1,2-Diones to Acetylenic Ester: Synthetic and Theoretical
Статья Paper. Denis V. Chachkov b and Oleg V. Mikhailov Теоретические исследования Theoretical Studies. Introduction. Method
 Теоретические исследования Theoretical Studies Статья Paper DFT B3LYP Quantum-Chemical Calculation of Molecular Structures of (6.6.6)Macrotricyclic M II Complexes with (N,N,N,N)-Coordinating Ligand Formed
Теоретические исследования Theoretical Studies Статья Paper DFT B3LYP Quantum-Chemical Calculation of Molecular Structures of (6.6.6)Macrotricyclic M II Complexes with (N,N,N,N)-Coordinating Ligand Formed
Final Report: Molecular simulation of copper(ii)-bound organic compounds for use in metalorganic chemical vapor deposition (MOCVD) of copper films
-bound organic compounds for use in metalorganic chemical vapor deposition (MOCVD) of copper films") Final Report: Molecular simulation of copper(ii)-bound organic compounds for use in metalorganic chemical vapor deposition (MOCVD) of copper films By: Rivera-Montalvo, Alexis A. University of Puerto Rico,
Final Report: Molecular simulation of copper(ii)-bound organic compounds for use in metalorganic chemical vapor deposition (MOCVD) of copper films By: Rivera-Montalvo, Alexis A. University of Puerto Rico,
ethers components: Ab initio study
 Arabian Journal of Chemistry (2016) 9, S240 S244 King Saud University Arabian Journal of Chemistry www.ksu.edu.sa www.sciencedirect.com ORIGINAL ARTICLE 17 O NMR parameters of some substituted benzyl ethers
Arabian Journal of Chemistry (2016) 9, S240 S244 King Saud University Arabian Journal of Chemistry www.ksu.edu.sa www.sciencedirect.com ORIGINAL ARTICLE 17 O NMR parameters of some substituted benzyl ethers
AN EXAMPLE REPORT. Cecil Dybowski List all names of people involved, along with addresses.
 AN EXAMPLE REPORT by Cecil Dybowski (dybowski@udel.edu) List all names of people involved, along with email addresses. CHEMISTRY 446 Section XL Here you enter the section and group numbers. EXPERIMENT
AN EXAMPLE REPORT by Cecil Dybowski (dybowski@udel.edu) List all names of people involved, along with email addresses. CHEMISTRY 446 Section XL Here you enter the section and group numbers. EXPERIMENT
China; University of Science and Technology, Nanjing , P R China.
 Electronic Supplementary Information Lithium-doped MOF impregnated with lithium-coated fullerenes: A hydrogen storage route for high gravimetric and volumetric uptakes at ambient temperatures Dewei Rao,
Electronic Supplementary Information Lithium-doped MOF impregnated with lithium-coated fullerenes: A hydrogen storage route for high gravimetric and volumetric uptakes at ambient temperatures Dewei Rao,
Photoinduced intramolecular charge transfer in trans-2-[4 -(N,Ndimethylamino)styryl]imidazo[4,5-b]pyridine:
![Photoinduced intramolecular charge transfer in trans-2-[4 -(N,Ndimethylamino)styryl]imidazo[4,5-b]pyridine:](/thumbs/74/70580684.jpg "Photoinduced intramolecular charge transfer in trans-2-[4 -(N,Ndimethylamino)styryl]imidazo[4,5-b]pyridine:") Electronic Supplementary Material (ESI) for Photochemical & Photobiological Sciences. This journal is The Royal Society of Chemistry and Owner Societies 2014 Photoinduced intramolecular charge transfer
Electronic Supplementary Material (ESI) for Photochemical & Photobiological Sciences. This journal is The Royal Society of Chemistry and Owner Societies 2014 Photoinduced intramolecular charge transfer
University of Groningen
 University of Groningen Tuning the Temperature Dependence for Switching in Dithienylethene Photochromic Switches Kudernac, Tibor; Kobayashi, Takao; Uyama, Ayaka; Uchida, Kingo; Nakamura, Shinichiro; Feringa,
University of Groningen Tuning the Temperature Dependence for Switching in Dithienylethene Photochromic Switches Kudernac, Tibor; Kobayashi, Takao; Uyama, Ayaka; Uchida, Kingo; Nakamura, Shinichiro; Feringa,
A Computational NMR Study of Boron Phosphide Nanotubes
 A Computational NMR Study of Boron Phosphide Nanotubes Mahmoud Mirzaei Young Researchers Club, Islamic Azad University, Shahr-e-Rey Branch, Shahr-e-Rey, Iran Reprint requests to M. M.; E-mail: mdmirzaei@yahoo.com
A Computational NMR Study of Boron Phosphide Nanotubes Mahmoud Mirzaei Young Researchers Club, Islamic Azad University, Shahr-e-Rey Branch, Shahr-e-Rey, Iran Reprint requests to M. M.; E-mail: mdmirzaei@yahoo.com
Supplementary Material
 The Electronic Spectrum of the C s -C 11 H 3 Radical Dongfeng Zhao, 1 Harold Linnartz,,1 and Wim Ubachs 1 1 Institute for Lasers, Life, and Biophotonics, VU University Amsterdam, De Boelelaan 1081, NL
The Electronic Spectrum of the C s -C 11 H 3 Radical Dongfeng Zhao, 1 Harold Linnartz,,1 and Wim Ubachs 1 1 Institute for Lasers, Life, and Biophotonics, VU University Amsterdam, De Boelelaan 1081, NL
The Chemist Journal of the American Institute of Chemists
 The Chemist Journal of the American Institute of Chemists Computational Studies on the IR and NMR Spectra of 2-Aminophenol Abraham George 1 *, P, V, Thomas 2, and David Devraj Kumar 3 1. Department of
The Chemist Journal of the American Institute of Chemists Computational Studies on the IR and NMR Spectra of 2-Aminophenol Abraham George 1 *, P, V, Thomas 2, and David Devraj Kumar 3 1. Department of
Supporting Information. for. Angew. Chem. Int. Ed. Z Wiley-VCH 2003
 Supporting Information for Angew. Chem. Int. Ed. Z52858 Wiley-VCH 23 69451 Weinheim, Germany Toward an Improved Understanding of the Unusual Reactivity of Pd()/Trialkylphosphine Catalysts in Cross-Couplings
Supporting Information for Angew. Chem. Int. Ed. Z52858 Wiley-VCH 23 69451 Weinheim, Germany Toward an Improved Understanding of the Unusual Reactivity of Pd()/Trialkylphosphine Catalysts in Cross-Couplings
1,5,2,4,6,8-dithiatetrazocine. Synthesis, computation, crystallography and voltammetry of the parent heterocycle. Supplemental Information
 1,5,2,4,6,8-dithiatetrazocine. Synthesis, computation, crystallography and voltammetry of the parent heterocycle. Klaus H. Moock 1, Ken M. Wong 2 and René T. Boeré* 2 Moock Environmental Solutions Ltd.,
1,5,2,4,6,8-dithiatetrazocine. Synthesis, computation, crystallography and voltammetry of the parent heterocycle. Klaus H. Moock 1, Ken M. Wong 2 and René T. Boeré* 2 Moock Environmental Solutions Ltd.,
Supporting Information. Synthesis, Molecular Structure, and Facile Ring Flipping of a Bicyclo[1.1.0]tetrasilane
![Supporting Information. Synthesis, Molecular Structure, and Facile Ring Flipping of a Bicyclo[1.1.0]tetrasilane](/thumbs/93/113883390.jpg "Supporting Information. Synthesis, Molecular Structure, and Facile Ring Flipping of a Bicyclo[1.1.0]tetrasilane") Supporting Information Synthesis, Molecular Structure, and Facile Ring Flipping of a Bicyclo[1.1.0]tetrasilane Kiyomi Ueba-Ohshima, Takeaki Iwamoto,*,# Mitsuo Kira*, #Research and Analytical Center for
Supporting Information Synthesis, Molecular Structure, and Facile Ring Flipping of a Bicyclo[1.1.0]tetrasilane Kiyomi Ueba-Ohshima, Takeaki Iwamoto,*,# Mitsuo Kira*, #Research and Analytical Center for
A theoretical study on the thermodynamic parameters for some imidazolium crystals
 Available online www.jocpr.com Journal of Chemical and Pharmaceutical Research, 2015, 7(2):550-554 Research Article ISSN : 0975-7384 CODEN(USA) : JCPRC5 A theoretical study on the thermodynamic parameters
Available online www.jocpr.com Journal of Chemical and Pharmaceutical Research, 2015, 7(2):550-554 Research Article ISSN : 0975-7384 CODEN(USA) : JCPRC5 A theoretical study on the thermodynamic parameters
Supporting Information For
 Supporting Information For Chemo-, Regio- and Stereoselective Synthesis of Polysusbtituted xazolo[3,2-d][1,4]oxazepin-5(3h)ones via a Domino oxa- Michael/aza-Michael/Williamson Cycloetherification Sequence
Supporting Information For Chemo-, Regio- and Stereoselective Synthesis of Polysusbtituted xazolo[3,2-d][1,4]oxazepin-5(3h)ones via a Domino oxa- Michael/aza-Michael/Williamson Cycloetherification Sequence
Ali Rostami, Alexis Colin, Xiao Yu Li, Michael G. Chudzinski, Alan J. Lough and Mark S. Taylor*
 N,N -Diaryl Squaramides: General, High-yielding Synthesis and Applications in Colorimetric Anion Sensing Ali Rostami, Alexis Colin, Xiao Yu Li, Michael G. Chudzinski, Alan J. Lough and Mark S. Taylor*
N,N -Diaryl Squaramides: General, High-yielding Synthesis and Applications in Colorimetric Anion Sensing Ali Rostami, Alexis Colin, Xiao Yu Li, Michael G. Chudzinski, Alan J. Lough and Mark S. Taylor*
Theoretical ab Initio Study the Hydrogen Bonding Nature of the A: T Base Pair
 International Journal of Pure and Applied Physics. ISSN 973-1776 Volume 7, Number 3 (211), pp. 251-261 Research India Publications http://www.ripublication.com/ijpap.htm Theoretical ab Initio Study the
International Journal of Pure and Applied Physics. ISSN 973-1776 Volume 7, Number 3 (211), pp. 251-261 Research India Publications http://www.ripublication.com/ijpap.htm Theoretical ab Initio Study the
SUPPLEMENTARY INFORMATION
 DOI: 10.1038/NCHEM.1754 Caesium in high oxidation states and as a p-block element Mao-sheng Miao Materials Research Laboratory, University of California, Santa Barbara, CA 93106-5050, USA and Beijing Computational
DOI: 10.1038/NCHEM.1754 Caesium in high oxidation states and as a p-block element Mao-sheng Miao Materials Research Laboratory, University of California, Santa Barbara, CA 93106-5050, USA and Beijing Computational
Computational Material Science Part II
 Computational Material Science Part II Ito Chao ( ) Institute of Chemistry Academia Sinica Aim of Part II Get familiar with the computational methodologies often used and properties often predicted in
Computational Material Science Part II Ito Chao ( ) Institute of Chemistry Academia Sinica Aim of Part II Get familiar with the computational methodologies often used and properties often predicted in
Which NICS Aromaticity Index for Planar π Rings is Best?
 S1 SUPPORTING INFORMATION Which NICS Aromaticity Index for Planar π Rings is Best? Hossein Fallah-Bagher-Shaidaei*,, Chaitanya S. Wannere, Clémence Corminboeuf, Ralph Puchta, and P. v. R. Schleyer *, Department
S1 SUPPORTING INFORMATION Which NICS Aromaticity Index for Planar π Rings is Best? Hossein Fallah-Bagher-Shaidaei*,, Chaitanya S. Wannere, Clémence Corminboeuf, Ralph Puchta, and P. v. R. Schleyer *, Department
Supporting Information
 Supporting Information Oxygen Atom Transfer Reactions of Iridium and Osmium Complexes: Theoretical Study of Characteristic Features and Significantly Large Differences Between These Two Complexes Atsushi
Supporting Information Oxygen Atom Transfer Reactions of Iridium and Osmium Complexes: Theoretical Study of Characteristic Features and Significantly Large Differences Between These Two Complexes Atsushi
STRUCTURAL DETERMINATION OF A SYNTHETIC POLYMER BY GAUSSIAN COMPUTATIONAL MODELING SOFTWARE
 STRUCTURAL DETERMINATIN F A SYNTHETIC PLYMER BY GAUSSIAN CMPUTATINAL MDELING SFTWARE AND NUCLEAR MAGNETIC RESNANCE SPECTRSCPY Kristen Entwistle*, Dwight Tshudy*, Terrence Collins** *Department of Chemistry,
STRUCTURAL DETERMINATIN F A SYNTHETIC PLYMER BY GAUSSIAN CMPUTATINAL MDELING SFTWARE AND NUCLEAR MAGNETIC RESNANCE SPECTRSCPY Kristen Entwistle*, Dwight Tshudy*, Terrence Collins** *Department of Chemistry,
Supporting Information
 Mechanical Control of ATP Synthase Function: Activation Energy Difference between Tight and Loose Binding Sites Supporting Information Tamás Beke-Somfai* 1,2, Per Lincoln 1, Bengt Nordén* 1 1 Department
Mechanical Control of ATP Synthase Function: Activation Energy Difference between Tight and Loose Binding Sites Supporting Information Tamás Beke-Somfai* 1,2, Per Lincoln 1, Bengt Nordén* 1 1 Department
Supporting Information. Copyright Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006
 Supporting Information Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2006 Formation and stability of G-quadruplex self-assembled from guanine-rich strands Jiang Zhou, Gu Yuan*, Junjun Liu,
Supporting Information Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2006 Formation and stability of G-quadruplex self-assembled from guanine-rich strands Jiang Zhou, Gu Yuan*, Junjun Liu,
Use of Ab Initio Calculations to Help Interpret the UV-Visible Spectra of Aquavanadium Complexes: A New Look at an Old Experiment 1
 Use of Ab Initio Calculations to Help Interpret the UV-Visible Spectra of Aquavanadium Complexes: A New Look at an Old Experiment 1 Wayne P. Anderson,* Department of Chemistry, Bloomsburg University of
Use of Ab Initio Calculations to Help Interpret the UV-Visible Spectra of Aquavanadium Complexes: A New Look at an Old Experiment 1 Wayne P. Anderson,* Department of Chemistry, Bloomsburg University of
(1) 2. Thermochemical calculations [2,3]
![(1) 2. Thermochemical calculations [2,3]](/thumbs/72/67567135.jpg "(1) 2. Thermochemical calculations [2,3]") 1. Introduction The exploration of reaction mechanisms and reaction paths that cannot be measured directly during an experiment has nowadays become a daily routine for chemists to support their laboratory
1. Introduction The exploration of reaction mechanisms and reaction paths that cannot be measured directly during an experiment has nowadays become a daily routine for chemists to support their laboratory
» ß π«õß 1 H-NMR 13 C-NMR ª ªï Õß
 « ß π«õß 1 H-NMR 13 C-NMR ª ªï Õß α-mangostin, γ-mangostin Garcinone D æ æ ª ß åæ π å* àõ ß π«π È ªìπ» ß π«à chemical shift Õß 1 H and 13 C NMR ÕßÕπÿæ π åÿ Õß ß µ π â à Õ ø - ß µ π - ß µ π å ππ â««wp04,
« ß π«õß 1 H-NMR 13 C-NMR ª ªï Õß α-mangostin, γ-mangostin Garcinone D æ æ ª ß åæ π å* àõ ß π«π È ªìπ» ß π«à chemical shift Õß 1 H and 13 C NMR ÕßÕπÿæ π åÿ Õß ß µ π â à Õ ø - ß µ π - ß µ π å ππ â««wp04,
A Redox-Fluorescent Molecular Switch Based on a. Heterobimetallic Ir(III) Complex with a Ferrocenyl. Azaheterocycle as Ancillary Ligand.
 Complex with a Ferrocenyl. Azaheterocycle as Ancillary Ligand.") Supporting Information (SI) A Redox-Fluorescent Molecular Switch Based on a Heterobimetallic Ir(III) Complex with a Ferrocenyl Azaheterocycle as Ancillary Ligand. Fabiola Zapata, Antonio Caballero, Arturo
Supporting Information (SI) A Redox-Fluorescent Molecular Switch Based on a Heterobimetallic Ir(III) Complex with a Ferrocenyl Azaheterocycle as Ancillary Ligand. Fabiola Zapata, Antonio Caballero, Arturo
Diphosphene Photobehaviour
 Electronic Supplementary Information P=P Bond Photophysics in an Ar-P=P-Ar Diphosphene Huo-Lei Peng, John L. Payton, John D. Protasiewicz, M. Cather Simpson Full references for Gaussian and MolPro. (9)
Electronic Supplementary Information P=P Bond Photophysics in an Ar-P=P-Ar Diphosphene Huo-Lei Peng, John L. Payton, John D. Protasiewicz, M. Cather Simpson Full references for Gaussian and MolPro. (9)
Concerted Attack of Frustrated Lewis Acid Base Pairs on Olefinic Double Bonds: A Theoretical Study
 Supporting Information Concerted Attack of Frustrated Lewis Acid Base Pairs on Olefinic Double Bonds: A Theoretical Study András Stirling, Andrea Hamza, Tibor András Rokob and Imre Pápai* Chemical Research
Supporting Information Concerted Attack of Frustrated Lewis Acid Base Pairs on Olefinic Double Bonds: A Theoretical Study András Stirling, Andrea Hamza, Tibor András Rokob and Imre Pápai* Chemical Research
Inverse Sodium Hydride: A Theoretical Study
 Published on Web 03/06/2003 Inverse Sodium Hydride: A Theoretical Study Agnieszka Sawicka,, Piotr Skurski,, and Jack Simons*, Contribution from the Henry Eyring Center for Theoretical Chemistry, Department
Published on Web 03/06/2003 Inverse Sodium Hydride: A Theoretical Study Agnieszka Sawicka,, Piotr Skurski,, and Jack Simons*, Contribution from the Henry Eyring Center for Theoretical Chemistry, Department
DFT Studies on HOMO-LUMO Gaps of CBNNTs
 DFT Studies on HOMO-LUMO Gaps of CBNNTs A. A. El-Barbary 1,2, Kh. M. Eid 1, M. A. Kamel 1, M. M. Hassan 1 1 Physics Department, Faculty of Education, Ain Shams University, Cairo, Egypt. 2 Physics Department,
DFT Studies on HOMO-LUMO Gaps of CBNNTs A. A. El-Barbary 1,2, Kh. M. Eid 1, M. A. Kamel 1, M. M. Hassan 1 1 Physics Department, Faculty of Education, Ain Shams University, Cairo, Egypt. 2 Physics Department,
Supporting Information
 A General Organocatalyst for Direct α-functionalization of Aldehydes: Stereoselective C-C, C-N, C-F, C-Br and C-S Bond-Forming Reactions. Scope and Mechanistic Insights Johan Franzén, Mauro Marigo, Doris
A General Organocatalyst for Direct α-functionalization of Aldehydes: Stereoselective C-C, C-N, C-F, C-Br and C-S Bond-Forming Reactions. Scope and Mechanistic Insights Johan Franzén, Mauro Marigo, Doris
Composition dependent properties of GaAs clusters
 Computer Physics Communications 142 (2001) 290 294 www.elsevier.com/locate/cpc Composition dependent properties of GaAs clusters H.H. Kwong, Y.P. Feng,T.B.Boo Department of Physics, National University
Computer Physics Communications 142 (2001) 290 294 www.elsevier.com/locate/cpc Composition dependent properties of GaAs clusters H.H. Kwong, Y.P. Feng,T.B.Boo Department of Physics, National University
Electronic Supplementary Information for:
 Electronic Supplementary Information for: The Potential of a cyclo-as 5 Ligand Complex in Coordination Chemistry H. Krauss, a G. Balazs, a M. Bodensteiner, a and M. Scheer* a a Institute of Inorganic Chemistry,
Electronic Supplementary Information for: The Potential of a cyclo-as 5 Ligand Complex in Coordination Chemistry H. Krauss, a G. Balazs, a M. Bodensteiner, a and M. Scheer* a a Institute of Inorganic Chemistry,
Supporting Online Material for
 Originally posted 2 July 2010; revised 4 March 2011 www.sciencemag.org/cgi/content/full/329/5987/65/dc1 Supporting Online Material for Does the Hydrated Electron Occupy a Cavity? Ross E. Larsen,* William
Originally posted 2 July 2010; revised 4 March 2011 www.sciencemag.org/cgi/content/full/329/5987/65/dc1 Supporting Online Material for Does the Hydrated Electron Occupy a Cavity? Ross E. Larsen,* William
Group 13 BN dehydrocoupling reagents, similar to transition metal catalysts but with unique reactivity. Part A: NMR Studies
 Part A: NMR Studies ESI 1 11 B NMR spectrum of the 2:1 reaction of i Pr 2 NHBH 3 with Al(NMe 2 ) 3 in d 6 -benzene 24 h later 11 B NMR ESI 2 11 B NMR spectrum of the reaction of t BuNH 2 BH 3 with Al(NMe
Part A: NMR Studies ESI 1 11 B NMR spectrum of the 2:1 reaction of i Pr 2 NHBH 3 with Al(NMe 2 ) 3 in d 6 -benzene 24 h later 11 B NMR ESI 2 11 B NMR spectrum of the reaction of t BuNH 2 BH 3 with Al(NMe
Supporting information
 Supporting information A Computational Study of the CO Dissociation in Cyclopentadienyl Ruthenium Complexes Relevant to the Racemization of Alcohols Beverly Stewart 1,2, Jonas Nyhlen 1, Belén Martín-Matute
Supporting information A Computational Study of the CO Dissociation in Cyclopentadienyl Ruthenium Complexes Relevant to the Racemization of Alcohols Beverly Stewart 1,2, Jonas Nyhlen 1, Belén Martín-Matute
A 3D-QSAR Study of Celebrex-based PDK1 Inhibitors Using CoMFA Method
 Journal of the Chinese Chemical Society, 2009, 56, 59-64 59 A 3D-QSAR Study of Celebrex-based PDK1 Inhibitors Using CoMFA Method Wen-Hung Wang a ( ),N.R.Jena a ( ), Yi-Ching Wang b ( ) and Ying-Chieh Sun
Journal of the Chinese Chemical Society, 2009, 56, 59-64 59 A 3D-QSAR Study of Celebrex-based PDK1 Inhibitors Using CoMFA Method Wen-Hung Wang a ( ),N.R.Jena a ( ), Yi-Ching Wang b ( ) and Ying-Chieh Sun
Supporting information on. Singlet Diradical Character from Experiment
 Supporting information on Singlet Diradical Character from Experiment Kenji Kamada,,* Koji Ohta, Akihiro Shimizu, Takashi Kubo,,* Ryohei Kishi, Hideaki Takahashi, Edith Botek, Benoît Champagne,,* and Masayoshi
Supporting information on Singlet Diradical Character from Experiment Kenji Kamada,,* Koji Ohta, Akihiro Shimizu, Takashi Kubo,,* Ryohei Kishi, Hideaki Takahashi, Edith Botek, Benoît Champagne,,* and Masayoshi
PERKIN. Theoretical characterization of 5-oxo, 7-oxo and 5-oxo-7- amino[1,2,4]triazolo[1,5-a]pyrimidines. I. Introduction. II.
![PERKIN. Theoretical characterization of 5-oxo, 7-oxo and 5-oxo-7- amino[1,2,4]triazolo[1,5-a]pyrimidines. I. Introduction. II.](/thumbs/92/110618854.jpg "PERKIN. Theoretical characterization of 5-oxo, 7-oxo and 5-oxo-7- amino[1,2,4]triazolo[1,5-a]pyrimidines. I. Introduction. II.") Theoretical characterization of 5-oxo, 7-oxo and 5-oxo-7- amino[1,2,4]triazolo[1,5-a]pyrimidines Jose A. Dobado,* Sonja Grigoleit and José Molina Molina Grupo de Modelización y Diseño Molecular, Instituto
Theoretical characterization of 5-oxo, 7-oxo and 5-oxo-7- amino[1,2,4]triazolo[1,5-a]pyrimidines Jose A. Dobado,* Sonja Grigoleit and José Molina Molina Grupo de Modelización y Diseño Molecular, Instituto
Supporting Information. A rare three-coordinated zinc cluster-organic framework
 Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 0 Supporting Information A rare three-coordinated zinc cluster-organic framework with two types of second
Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 0 Supporting Information A rare three-coordinated zinc cluster-organic framework with two types of second
Adsorption Properties of Oxygen on H-Capped (5, 5) Boron Nitride Nanotube (BNNT)- A Density Functional Theory
 Boron Nitride Nanotube (BNNT)- A Density Functional Theory") ISSN: 0973-4945; CODEN ECJHAO E- Chemistry http://www.e-journals.net 2011, 8(2), 609-614 Adsorption Properties of Oxygen on H-Capped (5, 5) Boron Nitride Nanotube (BNNT)- A Density Functional Theory MOHAMMAD
ISSN: 0973-4945; CODEN ECJHAO E- Chemistry http://www.e-journals.net 2011, 8(2), 609-614 Adsorption Properties of Oxygen on H-Capped (5, 5) Boron Nitride Nanotube (BNNT)- A Density Functional Theory MOHAMMAD
Electron Affinities of Selected Hydrogenated Silicon Clusters (Si x H y, x ) 1-7, y ) 0-15) from Density Functional Theory Calculations
 1-7, y ) 0-15) from Density Functional Theory Calculations") J. Phys. Chem. A 2000, 104, 6083-6087 6083 Electron Affinities of Selected Hydrogenated Silicon Clusters (Si x H y, x ) 1-7, y ) 0-15) from Density Functional Theory Calculations Mark T. Swihart Department
J. Phys. Chem. A 2000, 104, 6083-6087 6083 Electron Affinities of Selected Hydrogenated Silicon Clusters (Si x H y, x ) 1-7, y ) 0-15) from Density Functional Theory Calculations Mark T. Swihart Department
implications for different reactivity of identical subunits
 Supplementary Material Finding molecular dioxygen tunnels in homoprotocatechuate 2,3-dioxygenase: implications for different reactivity of identical subunits Liang Xu 1, 2, Weijie Zhao 3, Xicheng Wang
Supplementary Material Finding molecular dioxygen tunnels in homoprotocatechuate 2,3-dioxygenase: implications for different reactivity of identical subunits Liang Xu 1, 2, Weijie Zhao 3, Xicheng Wang
Supporting Information
 Supporting nformation Chromism Based on Supramolecular H-bonds Xiaowei Yu,, Chuanlang Zhan, *, Xunlei Ding, Shanlin Zhang, Xin Zhang, Huiying Liu, Lili Chen, Yishi Wu, Hongbing Fu, Shenggui He, *, Yan
Supporting nformation Chromism Based on Supramolecular H-bonds Xiaowei Yu,, Chuanlang Zhan, *, Xunlei Ding, Shanlin Zhang, Xin Zhang, Huiying Liu, Lili Chen, Yishi Wu, Hongbing Fu, Shenggui He, *, Yan
Supporting Information
 Supporting Information ucleophile-catalyzed Additions to Activated Triple Bonds. Protection of Lactams, Imides, and ucleosides with MocVinyl and Related Groups Laura Mola, Joan Font, Lluís Bosch, Joaquim
Supporting Information ucleophile-catalyzed Additions to Activated Triple Bonds. Protection of Lactams, Imides, and ucleosides with MocVinyl and Related Groups Laura Mola, Joan Font, Lluís Bosch, Joaquim
Supporting Information. Copyright Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006
 Supporting Information Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2006 ChemPhysChem Solvent effect on optical rotation: A case study of methyloxirane in water Parag Mukhopadhyay, Gérard
Supporting Information Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2006 ChemPhysChem Solvent effect on optical rotation: A case study of methyloxirane in water Parag Mukhopadhyay, Gérard
Phosphine Oxide Jointed Electron Transporters for Reducing Interfacial
 Electronic Supplementary Material (ESI) for Journal of Materials Chemistry C. This journal is The Royal Society of Chemistry 2015 Supporting Information Phosphine Oxide Jointed Electron Transporters for
Electronic Supplementary Material (ESI) for Journal of Materials Chemistry C. This journal is The Royal Society of Chemistry 2015 Supporting Information Phosphine Oxide Jointed Electron Transporters for
Ligand-to-Metal Ratio Controlled Assembly of Nanoporous Metal-Organic Frameworks
 Electronic Supplementary Information for Ligand-to-Metal Ratio Controlled Assembly of Nanoporous Metal-Organic Frameworks Jian-Guo Lin, a Yan-Yan Xu, a Ling Qiu, b Shuang-Quan Zang, a Chang-Sheng Lu, a
Electronic Supplementary Information for Ligand-to-Metal Ratio Controlled Assembly of Nanoporous Metal-Organic Frameworks Jian-Guo Lin, a Yan-Yan Xu, a Ling Qiu, b Shuang-Quan Zang, a Chang-Sheng Lu, a
Concerted halogen and hydrogen bonding in RuI 2 (H 2 dcbpy)(co) 2 ] I 2 (CH 3 OH) I 2 [RuI 2 (H 2 dcbpy)(co) 2 ]
![Concerted halogen and hydrogen bonding in RuI 2 (H 2 dcbpy)(co) 2 ] I 2 (CH 3 OH) I 2 [RuI 2 (H 2 dcbpy)(co) 2 ]](/thumbs/94/118476618.jpg "Concerted halogen and hydrogen bonding in RuI 2 (H 2 dcbpy)(co) 2 ] I 2 (CH 3 OH) I 2 [RuI 2 (H 2 dcbpy)(co) 2 ]") Concerted halogen and hydrogen bonding in RuI 2 (H 2 dcbpy)(co) 2 ] I 2 (CH 3 OH) I 2 [RuI 2 (H 2 dcbpy)(co) 2 ] Matti Tuikka a, Mika Niskanen a, Pipsa Hirva a, Kari Rissanen b, Arto Valkonen b, and Matti
Concerted halogen and hydrogen bonding in RuI 2 (H 2 dcbpy)(co) 2 ] I 2 (CH 3 OH) I 2 [RuI 2 (H 2 dcbpy)(co) 2 ] Matti Tuikka a, Mika Niskanen a, Pipsa Hirva a, Kari Rissanen b, Arto Valkonen b, and Matti
SUPPORTING INFORMATION
 SUPPORTING INFORMATION Revisiting the long-range Perlin effect in a conformationally constrained Oxocane Kahlil S. Salome and Cláudio F. Tormena* Institute of Chemistry, University of Campinas - UNICAMP
SUPPORTING INFORMATION Revisiting the long-range Perlin effect in a conformationally constrained Oxocane Kahlil S. Salome and Cláudio F. Tormena* Institute of Chemistry, University of Campinas - UNICAMP
This article is downloaded from.
 This article is downloaded from It is the paper published as: http://researchoutput.csu.edu.au Author: B. Kovac, K. Kowalski and I. Novak Title: Electronic Structure of 2,5-dimethylazaferrocene Journal:
This article is downloaded from It is the paper published as: http://researchoutput.csu.edu.au Author: B. Kovac, K. Kowalski and I. Novak Title: Electronic Structure of 2,5-dimethylazaferrocene Journal:
Solving multi-site conformational problems with total lineshape analysis of NMR spectral multiplets
 Solving multi-site conformational problems with total lineshape analysis of NMR spectral multiplets Sergei V. Zubkov, Sergei S. Golotvin, Vyacheslav A. Chertkov* Moscow State University, Department of
Solving multi-site conformational problems with total lineshape analysis of NMR spectral multiplets Sergei V. Zubkov, Sergei S. Golotvin, Vyacheslav A. Chertkov* Moscow State University, Department of
Molecular Modeling of Photoluminescent Copper(I) Cyanide Materials. Jasprina L Ming Advisor: Craig A Bayse
 Cyanide Materials. Jasprina L Ming Advisor: Craig A Bayse") Molecular Modeling of Photoluminescent Copper(I) Cyanide Materials Jasprina L Advisor: Craig A Bayse Department of Chemistry and Biochemistry, Old Dominion University, Hampton Boulevard, Norfolk, Virginia
Molecular Modeling of Photoluminescent Copper(I) Cyanide Materials Jasprina L Advisor: Craig A Bayse Department of Chemistry and Biochemistry, Old Dominion University, Hampton Boulevard, Norfolk, Virginia
Dynamics of H-atom loss in adenine: Supplementary information
 Dynamics of H-atom loss in adenine: Supplementary information M. Zierhut, W. Roth, and I. Fischer Institute of Physical Chemistry, University of Würzburg, Am Hubland, D-97074 Würzburg; Email: ingo@phys-chemie.uni-wuerzburg.de
Dynamics of H-atom loss in adenine: Supplementary information M. Zierhut, W. Roth, and I. Fischer Institute of Physical Chemistry, University of Würzburg, Am Hubland, D-97074 Würzburg; Email: ingo@phys-chemie.uni-wuerzburg.de
Reversible intercyclobutadiene haptotropism in cyclopentadienylcobalt linear [4]phenylene
![Reversible intercyclobutadiene haptotropism in cyclopentadienylcobalt linear [4]phenylene](/thumbs/95/123940099.jpg "Reversible intercyclobutadiene haptotropism in cyclopentadienylcobalt linear [4]phenylene") Reversible intercyclobutadiene haptotropism in cyclopentadienylcobalt linear [4]phenylene Thomas A. Albright, Sander Oldenhof, Oluwakemi A. Oloba, Robin Padilla and K. Peter C. Vollhardt * Experimental
Reversible intercyclobutadiene haptotropism in cyclopentadienylcobalt linear [4]phenylene Thomas A. Albright, Sander Oldenhof, Oluwakemi A. Oloba, Robin Padilla and K. Peter C. Vollhardt * Experimental
Journal of Physical and Theoretical Chemistry
 Journal of Physical and Theoretical Chemistry of Islamic Azad University of Iran, 8 (1) 1-9: Spring 2011 (J. Phys. Theor. Chem. IAU Iran) ISSN: 1735-2126 Ab initio Study of Simple -Ene Reactions of Propenyl
Journal of Physical and Theoretical Chemistry of Islamic Azad University of Iran, 8 (1) 1-9: Spring 2011 (J. Phys. Theor. Chem. IAU Iran) ISSN: 1735-2126 Ab initio Study of Simple -Ene Reactions of Propenyl
Supporting Information
 Missing Monometallofullerene with C 80 Cage Hidefumi Nikawa, Tomoya Yamada, Baopeng Cao, Naomi Mizorogi, Slanina Zdenek, Takahiro Tsuchiya, Takeshi Akasaka,* Kenji Yoza, Shigeru Nagase* Center for Tsukuba
Missing Monometallofullerene with C 80 Cage Hidefumi Nikawa, Tomoya Yamada, Baopeng Cao, Naomi Mizorogi, Slanina Zdenek, Takahiro Tsuchiya, Takeshi Akasaka,* Kenji Yoza, Shigeru Nagase* Center for Tsukuba
Ab Initio Calculations of 31 P NMR Chemical Shielding Anisotropy Tensors in Phosphates: Variations Due to Ring Formation
 Int. J. Mol. Sci. 00,, 888-90 International Journal of Molecular Sciences ISSN -007 00 by MDPI www.mdpi.org/ijms/ Ab Initio Calculations of P NMR Chemical Shielding Anisotropy Tensors in Phosphates: Variations
Int. J. Mol. Sci. 00,, 888-90 International Journal of Molecular Sciences ISSN -007 00 by MDPI www.mdpi.org/ijms/ Ab Initio Calculations of P NMR Chemical Shielding Anisotropy Tensors in Phosphates: Variations
Preprint. This is the submitted version of a paper published in Journal of Computational Chemistry.
 http://www.diva-portal.org Preprint This is the submitted version of a paper published in Journal of Computational Chemistry. Citation for the original published paper (version of record): Roca-Sanjuan,
http://www.diva-portal.org Preprint This is the submitted version of a paper published in Journal of Computational Chemistry. Citation for the original published paper (version of record): Roca-Sanjuan,
Electronic Supplementary Information (ESI) for Chem. Commun.
 for Chem. Commun.") page S1 Electronic Supplementary Information (ESI) for Chem. Commun. Nitric oxide coupling mediated by iron porphyrins: the N-N bond formation step is facilitated by electrons and a proton Jun Yi, Brian
page S1 Electronic Supplementary Information (ESI) for Chem. Commun. Nitric oxide coupling mediated by iron porphyrins: the N-N bond formation step is facilitated by electrons and a proton Jun Yi, Brian