Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics
|
|
|
- Spencer Eaton
- 5 years ago
- Views:
Transcription
1 Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics Tandy Warnow Founder Professor of Engineering The University of Illinois at Urbana-Champaign




2 From the Tree of the Life Website, University of Arizona Phylogenomics






 Gene Tree")
3 1kp: Thousand Transcriptome Project G. Ka-Shu Wong U Alberta J. Leebens-Mack U Georgia N. Wickett Northwestern N. Matasci iplant T. Warnow, UIUC S. Mirarab, UCSD N. Nguyen UCSD Plus many many other people Plant Tree of Life based on transcriptomes of ~1200 species l More than 13,000 gene families (most not single copy) Gene Tree Incongruence l Challenge: Alignments and trees on > 100,000 sequences
4 1000-taxon models, ordered by difficulty (Liu et al., Science 324(5934): , 2009)

5 Re-aligning on a tree A B C D Decompose dataset A C B D Align subsets A C B D Estimate ML tree on merged alignment ABCD Merge sub-alignments
6 SATé and PASTA Algorithms Obtain initial alignment and estimated ML tree Estimate ML tree on new alignment Tree Use tree to compute new alignment Alignment Repeat until termination condition, and return the alignment/tree pair with the best ML score
7 1000 taxon models, ordered by difficulty, Liu et al., Science 324(5934): , hour SATé-I analysis, on desktop machines (Similar improvements for biological datasets)
8 SATé-2 better than SATé taxon models ranked by difficulty SATé-1 (Liu et al., Science 2009): can analyze up to 8K sequences SATé-2 (Liu et al., Systematic Biology 2012): can analyze up to ~50K sequences
9 PASTA: even better than SATé-2 RNASim 0.20 Tree Error (FN Rate) Clustal Omega Muscle Mafft Starting Tree SATe2 PASTA Reference Alignment PASTA: Mirarab, Nguyen, and Warnow, J Comp. Biol Simulated RNASim datasets from 10K to 200K taxa Limited to 24 hours using 12 CPUs Not all methods could run (missing bars could not finish)
10 0.4 Total Column Score PASTA+BAli Phy Better Comparing default PASTA to PASTA+BAli-Phy on simulated datasets with 1000 sequences PASTA+BAli Phy PASTA Better PASTA data Indelible M2 RNAsim Rose L1 Rose M1 Rose S1 Recall (SP Score) Tree Error: Delta RF (RAxML) 1.0 PASTA+BAli Phy Better 0.15 PASTA+BAli Phy Better PASTA+BAli Phy PASTA Better PASTA PASTA Better PASTA PASTA+BAli Phy Decomposition to 100-sequence subsets, one iteration of PASTA+BAli-Phy
11 1kp: Thousand Transcriptome Project G. Ka-Shu Wong U Alberta J. Leebens-Mack U Georgia N. Wickett Northwestern N. Matasci iplant T. Warnow, UIUC S. Mirarab, UCSD N. Nguyen UCSD Plus many many other people Plant Tree of Life based on transcriptomes of ~1200 species l More than 13,000 gene families (most not single copy) Gene Tree Incongruence l Challenge: Alignments and trees on > 100,000 sequences
12 12000 Mean: Median:266 1KP dataset: more than 100,000 p450 amino-acid sequences, many fragmentary 8000 Counts Length
13 12000 Mean: Median:266 1KP dataset: more than 100,000 p450 amino-acid sequences, many fragmentary 8000 Counts All standard multiple sequence alignment methods we tested performed poorly on datasets with fragments Length
14 Profile Hidden Markov Models Probabilistic model to represent a family of sequences, represented by a multiple sequence alignment Introduced for sequence analysis in Krogh et al Popularized in Eddy 1996 and textbook Durbin et al Fundamental part of HMMER and other protein databases Used for: homology detection, protein family assignment, multiple sequence alignment, phylogenetic placement, protein structure prediction, alignment segmentation, etc.
15 Profile HMMs l l Generative model for representing a MSA Consists of: l l l Set of states (Match, insertion, and deletion) Transition probabilities Emission probabilities
16 Profile Hidden Markov Model for DNA sequence alignment
17 HMMs for MSA l l Given seed alignment (e.g., in PFAM) and a collection of sequences for the protein family: l l l Represent seed alignment using HMM Align each additional sequence to the HMM Use transitivity to obtain MSA Can we do something like this without a seed alignment?
18 HMMs for MSA l l Given seed alignment (e.g., in PFAM) and a collection of sequences for the protein family: l l l Represent seed alignment using HMM Align each additional sequence to the HMM Use transitivity to obtain MSA Can we do something like this without a seed alignment?
19 UPP UPP = Ultra-large multiple sequence alignment using Phylogeny-aware Profiles Nguyen, Mirarab, and Warnow. Genome Biology, Purpose: highly accurate large-scale multiple sequence alignments, even in the presence of fragmentary sequences.
20 UPP UPP = Ultra-large multiple sequence alignment using Phylogeny-aware Profiles Nguyen, Mirarab, and Warnow. Genome Biology, Purpose: highly accurate large-scale multiple sequence alignments, even in the presence of fragmentary sequences. Uses an ensemble of HMMs
21 Simple idea (not UPP) Select random subset of sequences, and build backbone alignment Construct a Hidden Markov Model (HMM) on the backbone alignment Add all remaining sequences to the backbone alignment using the HMM
22 PASTA: even better than SATé RNASim Starting tree is based on UPP(simple): one profile HMM for Backbone alignment Tree Error (FN Rate) Clustal Omega Muscle Mafft Starting Tree SATe2 PASTA Reference Alignment PASTA: Mirarab, Nguyen, and Warnow, J Comp. Biol Simulated RNASim datasets from 10K to 200K taxa Limited to 24 hours using 12 CPUs Not all methods could run (missing bars could not finish)
23 This approach works well if the dataset is small and has low evolutionary rates, but is not very accurate otherwise. Select random subset of sequences, and build backbone alignment Construct a Hidden Markov Model (HMM) on the backbone alignment Add all remaining sequences to the backbone alignment using the HMM
24 One Hidden Markov Model for the entire alignment?
25 One Hidden Markov Model for the backbone alignment? HMM 1
26 Or 2 HMMs? HMM 1 HMM 2
27 Or 4 HMMs? HMM 1 HMM 2 HMM 3 HMM 4
28 Or all 7 HMMs? HMM 1 HMM 2 HMM 4 m HMM 7 HMM 3 HMM 5 HMM 6
29 UPP Algorithmic Approach 1. Select random subset of full-length sequences, and build backbone alignment 2. Construct an Ensemble of Hidden Markov Models on the backbone alignment 3. Add all remaining sequences to the backbone alignment using the Ensemble of HMMs
30 Evaluation Simulated datasets (some have fragmentary sequences): 10K to 1,000,000 sequences in RNASim complex RNA sequence evolution simulation 1000-sequence nucleotide datasets from SATé papers 5000-sequence AA datasets (from FastTree paper) 10,000-sequence Indelible nucleotide simulation Biological datasets: Proteins: largest BaliBASE and HomFam RNA: 3 CRW datasets up to 28,000 sequences
 took 2.2 days, UPP(Fast) took 11.")
31 RNASim Million Sequences: tree error Using 12 processors: UPP(Fast,NoDecomp) took 2.2 days, UPP(Fast) took 11.9 days, and PASTA took 10.3 days
32 UPP is very robust to fragmentary sequences Mean alignment error Delta FN tree error % Fragmentary PASTA UPP(Default) (a) Average alignment error % Fragmentary Under high rates of evolution, PASTA is badly impacted by fragmentary sequences (the same is true for other methods). UPP continues to have good accuracy even on datasets with many fragments under all rates of evolution. PASTA UPP(Default) (b) Average tree error Performance on fragmentary datasets of the 1000M2 model condition
33 UPP Running Time Wall clock align time (hr) Number of sequences UPP(Fast) Wall-clock time used (in hours) given 12 processors
34 Other Applications of the Ensemble of HMMs SEPP (phylogenetic placement, Mirarab, Nguyen, and Warnow PSB 2014) TIPP (metagenomic taxon identification, Nguyen, Mirarab, Liu, Pop, and Warnow, Bioinformatics 2014) HIPPI (protein classification and remote homology detection), RECOMB-CG 2016 and BMC Genomics 2016 (Nguyen, Nute, Mirarab, and Warnow)
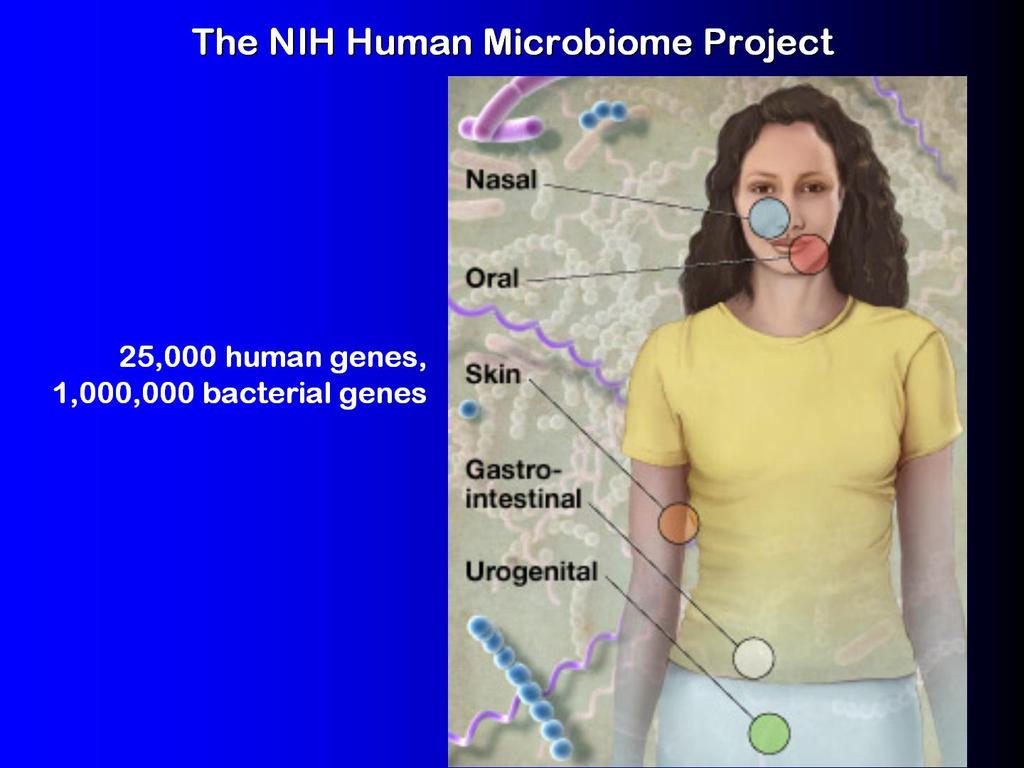
35
36 Abundance Profiling Objective: Distribution of the species (or genera, or families, etc.) within the sample. For example: The distribution of the sample at the species-level is: 50% species A 20% species B 15% species C 14% species D 1% species E
37 TIPP pipeline Input: set of reads from a shotgun sequencing experiment of a metagenomic sample 1. Assign reads to marker genes using BLAST 2. For reads assigned to marker genes, perform taxonomic analysis 3. Combine analyses from Step 2

38 High indel datasets containing known genomes Note: NBC, MetaPhlAn, and MetaPhyler cannot classify any sequences from at least one of the high indel long sequence datasets, and motu terminates with an error message on all the high indel datasets.

39 Novel genome datasets Note: motu terminates with an error message on the long fragment datasets and high indel datasets.
40 Protein Family Assignment Input: new AA sequence (might be fragmentary) and database of protein families (e.g., PFAM) Output: assignment (if justified) of the sequence to an existing family in the database
41 HIPPI HIerarchical Profile HMMs for Protein family Identification Nguyen, Nute, Mirarab, and Warnow, RECOMB-CG 2016 and BMC-Genomics 2016 Uses an ensemble of HMMs to classify protein sequences Tested on HMMER
42 Seq Length: Full Seq Length: 50% Seq Length: 25% Precision Recall Method HIPPI HMMER BLAST HHsearch: 1 Iteration HHsearch: 2 Iterations
43 Four Problems Phylogenetic Placement (SEPP, PSB 2012) Multiple sequence alignment (UPP, RECOMB 2014 and Genome Biology 2014) Metagenomic taxon identification (TIPP, Bioinformatics 2014) Gene family assignment and homology detection (HIPPI, RECOMB-CG 2016 and BMC Genomics 2016) A unifying technique is the Ensemble of Hidden Markov Models (introduced by Mirarab et al., 2012)
44 Summary Using an ensemble of HMMs tends to improve accuracy, for a cost of running time. Applications so far to taxonomic placement (SEPP), multiple sequence alignment (UPP), protein family classification (HIPPI). Improvements are mostly noticeable for large diverse datasets. Phylogenetically-based construction of the ensemble helps accuracy (note: the decompositions we produce are not clade-based), but the design and use of these ensembles is still in its infancy. (Many relatively similar approaches have been used by others, including Sci-Phy and FlowerPower by Sjolander.) The basic idea can be used with any kind of probabilistic model, doesn t have to be restricted to profile HMMs.
45 Basic question: why does it help? Using an ensemble of HMMs tends to improve accuracy, for a cost of running time. Applications so far to taxonomic placement (SEPP), multiple sequence alignment (UPP), protein family classification (HIPPI). Improvements are mostly noticeable for large diverse datasets. Phylogenetically-based construction of the ensemble helps accuracy (note: the decompositions we produce are not clade-based), but the design and use of these ensembles is still in its infancy. (Many relatively similar approaches have been used by others, including Sci-Phy and FlowerPower by Sjolander.) The basic idea can be used with any kind of probabilistic model, doesn t have to be restricted to profile HMMs.

46 The Tree of Life: Multiple Challenges Scientific challenges: Ultra-large multiple-sequence alignment Alignment-free phylogeny estimation Supertree estimation Estimating species trees from many gene trees Genome rearrangement phylogeny Reticulate evolution Visualization of large trees and alignments Data mining techniques to explore multiple optima Theoretical guarantees under Markov models of evolution Techniques: machine learning, applied probability theory, graph theory, combinatorial optimization, supercomputing, and heuristics
 and Siavash Mirarab (now faculty at UCSD), undergrad:")
 Current NSF grants: ABI-1458652 (multiple")


47 Acknowledgments PASTA and UPP: Nam Nguyen (now postdoc at UIUC) and Siavash Mirarab (now faculty at UCSD), undergrad: Keerthana Kumar (at UT-Austin) PASTA+BAli-Phy: Mike Nute (PhD student at UIUC) Current NSF grants: ABI (multiple sequence alignment) Grainger Foundation (at UIUC), and UIUC TACC, UTCS, Blue Waters, and UIUC campus cluster PASTA, UPP, SEPP, and TIPP are available on github at see also PASTA+BAli-Phy at
48 Nguyen et al. Genome Biology (2015) 16:124 Page 6 of 15 Table 2 Average alignment SP-error, tree error, and TC score across most full-length datasets Method ROSE RNASim Indelible ROSE CRW 10 AA HomFam HomFam NT 10K 10K AA (17) (2) Average alignment SP-error UPP 7.8 (1) 9.5 (1) 1.7 (2) 2.9 (1) 12.5 (1) 24.2 (1) 23.3 (1) 20.8 (2) PASTA 7.8 (1) 15.0 (2) 0.4 (1) 3.1 (1) 12.8 (1) 24.0 (1) 22.5 (1) 17.3 (1) MAFFT 20.6 (2) 25.5 (3) 41.4 (3) 4.9 (2) 28.3 (2) 23.5 (1) 25.3 (2) 20.7 (2) Muscle 20.6 (2) 64.7 (5) 62.4 (4) 5.5 (3) 30.7 (3) 30.2 (2) 48.1 (4) X Clustal 49.2 (3) 35.3 (4) X 6.5 (4) 43.3 (4) 24.3 (1) 27.7 (3) 29.4 (3) Average FN error UPP 1.3 (1) 0.8 (1) 0.3 (1) 1.8 (1) 7.8 (2) 3.4 (2) NA NA PASTA 1.3 (1) 0.4 (1) <0.1 (1) 1.3 (1) 5.1 (1) 3.3 (1) NA NA MAFFT 5.8 (2) 3.5 (2) 24.8 (3) 4.5 (3) 10.1 (3) 2.3 (1) NA NA Muscle 8.4 (3) 7.3 (3) 32.5 (4) 3.1 (2) 5.5 (1) 12.6 (3) NA NA Clustal 24.3 (4) 10.4 (4) X 4.2 (3) 34.1 (4) 3.5 (2) NA NA Average TC score UPP 37.8 (1) 0.5 (2) 11.0 (3) 2.6 (2) 1.4 (1) 11.4 (1) 47.3 (1) 40.3 (3) PASTA 37.8 (1) 2.3 (1) 48.0 (1) 5.4 (1) 2.3 (1) 12.1 (1) 46.1 (2) 50.0 (1) MAFFT 31.4 (2) 0.4 (2) 7.8 (4) 0.6 (3) 0.7 (2) 12.1 (1) 45.5 (2) 46.9 (2) Muscle 9.8 (3) <0.0 (2) 18.3 (2) 2.7 (2) 0.7 (2) 10.5 (2) 27.7 (4) X Clustal 5.7 (4) 0.2 (2) X 3.1 (2) 0.1 (2) 11.8 (1) 38.6 (3) 31.0 (4) We report the average alignment SP-error (the average of SPFN and SPFP errors) (top), average FN error (middle), and average TC score (bottom), for the collection of full-length datasets. All scores represent percentages and so are out of 100. Results marked with an X indicate that the method failed to terminate withinthe time limit (24 hours on a 12-core machine). Muscle failed to align two of the HomFam datasets; we report separate average results on the 17 HomFam datasets for all methods and the two HomFam datasets for all but Muscle. We did not test tree error on the HomFam datasets (therefore, the FN error is indicated by NA ). The tier ranking for each method is shown parenthetically
49 Precision Avg. Pairwise Sequence Identiy > 30% 20 30% < 20% Size: Size: > 100 Size: Size: > 100 Size: Size: > 100 Sequence Length: Full Sequence Length: 50% Sequence Length: 25% Recall Method HIPPI HMMER BLAST HHsearch: 1 Iteration HHsearch: 2 Iterations
50 1. Pre-processing seed alignments Family A Family B Family N i) Compute an ML tree on each seed alignment ii) Build ensemble of HMMs for each seed alignment, using its ML tree HMM A 1 HMM A3 HMM A2 HMM N 1 HMM N4 HMM B 1 HMM N3 HMM N 5 iii) Collect HMMs into database HMM database HMM N 2 HMM A 1 HMM A2 HMM A3 HMM B 1... HMM N 1 HMM N 2 HMM N3 HMM N4 HMM N 5 2. Classification of query sequences Query sequence HMM database HMM A 1 HMM A2 HMM A3 HMM B 1... HMM N 1 HMM N 2 HMM N3 HMM N4 HMM N 5 i) Score query sequence against all HMMs in database, keeping only scores above inclusion threshold HMM A1 = 8.9 HMM A2 = 7.3 HMM A3 = 9.4 HMM B1 = HMM N1 = 4.4 HMM N2 = 5.6 HMM N3 = 4.3 HMM N4 = 4.7 HMM N5 = 6.6 ii) Rank families by best scoring HMM within family 1) Family A = 9.4 2) Family N = ) Family B = iii) Assign query sequence to top ranking family Family A Query
Phylogenomics, Multiple Sequence Alignment, and Metagenomics. Tandy Warnow University of Illinois at Urbana-Champaign
 Phylogenomics, Multiple Sequence Alignment, and Metagenomics Tandy Warnow University of Illinois at Urbana-Champaign Phylogeny (evolutionary tree) Orangutan Gorilla Chimpanzee Human From the Tree of the
Phylogenomics, Multiple Sequence Alignment, and Metagenomics Tandy Warnow University of Illinois at Urbana-Champaign Phylogeny (evolutionary tree) Orangutan Gorilla Chimpanzee Human From the Tree of the
CS 581 Algorithmic Computational Genomics. Tandy Warnow University of Illinois at Urbana-Champaign
 CS 581 Algorithmic Computational Genomics Tandy Warnow University of Illinois at Urbana-Champaign Today Explain the course Introduce some of the research in this area Describe some open problems Talk about
CS 581 Algorithmic Computational Genomics Tandy Warnow University of Illinois at Urbana-Champaign Today Explain the course Introduce some of the research in this area Describe some open problems Talk about
Ultra- large Mul,ple Sequence Alignment
 Ultra- large Mul,ple Sequence Alignment Tandy Warnow Founder Professor of Engineering The University of Illinois at Urbana- Champaign hep://tandy.cs.illinois.edu Phylogeny (evolu,onary tree) Orangutan
Ultra- large Mul,ple Sequence Alignment Tandy Warnow Founder Professor of Engineering The University of Illinois at Urbana- Champaign hep://tandy.cs.illinois.edu Phylogeny (evolu,onary tree) Orangutan
Construc)ng the Tree of Life: Divide-and-Conquer! Tandy Warnow University of Illinois at Urbana-Champaign
ng the Tree of Life: Divide-and-Conquer! Tandy Warnow University of Illinois at Urbana-Champaign") Construc)ng the Tree of Life: Divide-and-Conquer! Tandy Warnow University of Illinois at Urbana-Champaign Phylogeny (evolutionary tree) Orangutan Gorilla Chimpanzee Human From the Tree of the Life Website,
Construc)ng the Tree of Life: Divide-and-Conquer! Tandy Warnow University of Illinois at Urbana-Champaign Phylogeny (evolutionary tree) Orangutan Gorilla Chimpanzee Human From the Tree of the Life Website,
CS 394C Algorithms for Computational Biology. Tandy Warnow Spring 2012
 CS 394C Algorithms for Computational Biology Tandy Warnow Spring 2012 Biology: 21st Century Science! When the human genome was sequenced seven years ago, scientists knew that most of the major scientific
CS 394C Algorithms for Computational Biology Tandy Warnow Spring 2012 Biology: 21st Century Science! When the human genome was sequenced seven years ago, scientists knew that most of the major scientific
SEPP and TIPP for metagenomic analysis. Tandy Warnow Department of Computer Science University of Texas
 SEPP and TIPP for metagenomic analysis Tandy Warnow Department of Computer Science University of Texas Phylogeny (evolutionary tree) Orangutan From the Tree of the Life Website, University of Arizona Gorilla
SEPP and TIPP for metagenomic analysis Tandy Warnow Department of Computer Science University of Texas Phylogeny (evolutionary tree) Orangutan From the Tree of the Life Website, University of Arizona Gorilla
SEPP and TIPP for metagenomic analysis. Tandy Warnow Department of Computer Science University of Texas
 SEPP and TIPP for metagenomic analysis Tandy Warnow Department of Computer Science University of Texas Metagenomics: Venter et al., Exploring the Sargasso Sea: Scientists Discover One Million New Genes
SEPP and TIPP for metagenomic analysis Tandy Warnow Department of Computer Science University of Texas Metagenomics: Venter et al., Exploring the Sargasso Sea: Scientists Discover One Million New Genes
NJMerge: A generic technique for scaling phylogeny estimation methods and its application to species trees
 NJMerge: A generic technique for scaling phylogeny estimation methods and its application to species trees Erin Molloy and Tandy Warnow {emolloy2, warnow}@illinois.edu University of Illinois at Urbana
NJMerge: A generic technique for scaling phylogeny estimation methods and its application to species trees Erin Molloy and Tandy Warnow {emolloy2, warnow}@illinois.edu University of Illinois at Urbana
CS 581 Algorithmic Computational Genomics. Tandy Warnow University of Illinois at Urbana-Champaign
 CS 581 Algorithmic Computational Genomics Tandy Warnow University of Illinois at Urbana-Champaign Course Staff Professor Tandy Warnow Office hours Tuesdays after class (2-3 PM) in Siebel 3235 Email address:
CS 581 Algorithmic Computational Genomics Tandy Warnow University of Illinois at Urbana-Champaign Course Staff Professor Tandy Warnow Office hours Tuesdays after class (2-3 PM) in Siebel 3235 Email address:
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Mul$ple Sequence Alignment Methods. Tandy Warnow Departments of Bioengineering and Computer Science h?p://tandy.cs.illinois.edu
 Mul$ple Sequence Alignment Methods Tandy Warnow Departments of Bioengineering and Computer Science h?p://tandy.cs.illinois.edu Species Tree Orangutan Gorilla Chimpanzee Human From the Tree of the Life
Mul$ple Sequence Alignment Methods Tandy Warnow Departments of Bioengineering and Computer Science h?p://tandy.cs.illinois.edu Species Tree Orangutan Gorilla Chimpanzee Human From the Tree of the Life
SUPPLEMENTARY INFORMATION
 Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Bioinformatics tools for phylogeny and visualization. Yanbin Yin
 Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
From Genes to Genomes and Beyond: a Computational Approach to Evolutionary Analysis. Kevin J. Liu, Ph.D. Rice University Dept. of Computer Science
 From Genes to Genomes and Beyond: a Computational Approach to Evolutionary Analysis Kevin J. Liu, Ph.D. Rice University Dept. of Computer Science!1 Adapted from U.S. Department of Energy Genomic Science
From Genes to Genomes and Beyond: a Computational Approach to Evolutionary Analysis Kevin J. Liu, Ph.D. Rice University Dept. of Computer Science!1 Adapted from U.S. Department of Energy Genomic Science
InDel 3-5. InDel 8-9. InDel 3-5. InDel 8-9. InDel InDel 8-9
 Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
PHYLOGENY AND SYSTEMATICS
 AP BIOLOGY EVOLUTION/HEREDITY UNIT Unit 1 Part 11 Chapter 26 Activity #15 NAME DATE PERIOD PHYLOGENY AND SYSTEMATICS PHYLOGENY Evolutionary history of species or group of related species SYSTEMATICS Study
AP BIOLOGY EVOLUTION/HEREDITY UNIT Unit 1 Part 11 Chapter 26 Activity #15 NAME DATE PERIOD PHYLOGENY AND SYSTEMATICS PHYLOGENY Evolutionary history of species or group of related species SYSTEMATICS Study
Grundlagen der Bioinformatik Summer semester Lecturer: Prof. Daniel Huson
 Grundlagen der Bioinformatik, SS 10, D. Huson, April 12, 2010 1 1 Introduction Grundlagen der Bioinformatik Summer semester 2010 Lecturer: Prof. Daniel Huson Office hours: Thursdays 17-18h (Sand 14, C310a)
Grundlagen der Bioinformatik, SS 10, D. Huson, April 12, 2010 1 1 Introduction Grundlagen der Bioinformatik Summer semester 2010 Lecturer: Prof. Daniel Huson Office hours: Thursdays 17-18h (Sand 14, C310a)
Multiple sequence alignment
 Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
CONCEPT OF SEQUENCE COMPARISON. Natapol Pornputtapong 18 January 2018
 CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
BLAST. Varieties of BLAST
 BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
From Gene Trees to Species Trees. Tandy Warnow The University of Texas at Aus<n
 From Gene Trees to Species Trees Tandy Warnow The University of Texas at Aus
From Gene Trees to Species Trees Tandy Warnow The University of Texas at Aus
SUPPLEMENTARY INFORMATION
 Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
08/21/2017 BLAST. Multiple Sequence Alignments: Clustal Omega
 BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Chapter 26: Phylogeny and the Tree of Life Phylogenies Show Evolutionary Relationships
 Chapter 26: Phylogeny and the Tree of Life You Must Know The taxonomic categories and how they indicate relatedness. How systematics is used to develop phylogenetic trees. How to construct a phylogenetic
Chapter 26: Phylogeny and the Tree of Life You Must Know The taxonomic categories and how they indicate relatedness. How systematics is used to develop phylogenetic trees. How to construct a phylogenetic
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
PROTEIN FUNCTION PREDICTION WITH AMINO ACID SEQUENCE AND SECONDARY STRUCTURE ALIGNMENT SCORES
 PROTEIN FUNCTION PREDICTION WITH AMINO ACID SEQUENCE AND SECONDARY STRUCTURE ALIGNMENT SCORES Eser Aygün 1, Caner Kömürlü 2, Zafer Aydin 3 and Zehra Çataltepe 1 1 Computer Engineering Department and 2
PROTEIN FUNCTION PREDICTION WITH AMINO ACID SEQUENCE AND SECONDARY STRUCTURE ALIGNMENT SCORES Eser Aygün 1, Caner Kömürlü 2, Zafer Aydin 3 and Zehra Çataltepe 1 1 Computer Engineering Department and 2
Dr. Amira A. AL-Hosary
 Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Bioinformatics. Dept. of Computational Biology & Bioinformatics
 Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Protein Bioinformatics. Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet sandberg.cmb.ki.
 Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools. Giri Narasimhan
 CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs15.html Describing & Modeling Patterns
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs15.html Describing & Modeling Patterns
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut
 Faculty of Veterinary Medicine Assiut") Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
Gibbs Sampling Methods for Multiple Sequence Alignment
 Gibbs Sampling Methods for Multiple Sequence Alignment Scott C. Schmidler 1 Jun S. Liu 2 1 Section on Medical Informatics and 2 Department of Statistics Stanford University 11/17/99 1 Outline Statistical
Gibbs Sampling Methods for Multiple Sequence Alignment Scott C. Schmidler 1 Jun S. Liu 2 1 Section on Medical Informatics and 2 Department of Statistics Stanford University 11/17/99 1 Outline Statistical
MiGA: The Microbial Genome Atlas
 December 12 th 2017 MiGA: The Microbial Genome Atlas Jim Cole Center for Microbial Ecology Dept. of Plant, Soil & Microbial Sciences Michigan State University East Lansing, Michigan U.S.A. Where I m From
December 12 th 2017 MiGA: The Microbial Genome Atlas Jim Cole Center for Microbial Ecology Dept. of Plant, Soil & Microbial Sciences Michigan State University East Lansing, Michigan U.S.A. Where I m From
Similarity searching summary (2)
") Similarity searching / sequence alignment summary Biol4230 Thurs, February 22, 2016 Bill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 What have we covered? Homology excess similiarity but no excess similarity
Similarity searching / sequence alignment summary Biol4230 Thurs, February 22, 2016 Bill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 What have we covered? Homology excess similiarity but no excess similarity
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Phylogenies Scores for Exhaustive Maximum Likelihood and Parsimony Scores Searches
 Int. J. Bioinformatics Research and Applications, Vol. x, No. x, xxxx Phylogenies Scores for Exhaustive Maximum Likelihood and s Searches Hyrum D. Carroll, Perry G. Ridge, Mark J. Clement, Quinn O. Snell
Int. J. Bioinformatics Research and Applications, Vol. x, No. x, xxxx Phylogenies Scores for Exhaustive Maximum Likelihood and s Searches Hyrum D. Carroll, Perry G. Ridge, Mark J. Clement, Quinn O. Snell
CISC 636 Computational Biology & Bioinformatics (Fall 2016)
") CISC 636 Computational Biology & Bioinformatics (Fall 2016) Predicting Protein-Protein Interactions CISC636, F16, Lec22, Liao 1 Background Proteins do not function as isolated entities. Protein-Protein
CISC 636 Computational Biology & Bioinformatics (Fall 2016) Predicting Protein-Protein Interactions CISC636, F16, Lec22, Liao 1 Background Proteins do not function as isolated entities. Protein-Protein
Phylogenetic Analysis. Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center
 Phylogenetic Analysis Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center Outline Basic Concepts Tree Construction Methods Distance-based methods
Phylogenetic Analysis Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center Outline Basic Concepts Tree Construction Methods Distance-based methods
Sara C. Madeira. Universidade da Beira Interior. (Thanks to Ana Teresa Freitas, IST for useful resources on this subject)
") Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
O 3 O 4 O 5. q 3. q 4. Transition
 Hidden Markov Models Hidden Markov models (HMM) were developed in the early part of the 1970 s and at that time mostly applied in the area of computerized speech recognition. They are first described in
Hidden Markov Models Hidden Markov models (HMM) were developed in the early part of the 1970 s and at that time mostly applied in the area of computerized speech recognition. They are first described in
ASTRAL: Fast coalescent-based computation of the species tree topology, branch lengths, and local branch support
 ASTRAL: Fast coalescent-based computation of the species tree topology, branch lengths, and local branch support Siavash Mirarab University of California, San Diego Joint work with Tandy Warnow Erfan Sayyari
ASTRAL: Fast coalescent-based computation of the species tree topology, branch lengths, and local branch support Siavash Mirarab University of California, San Diego Joint work with Tandy Warnow Erfan Sayyari
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT
 5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
Effects of Gap Open and Gap Extension Penalties
 Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Microbial analysis with STAMP
 Microbial analysis with STAMP Conor Meehan cmeehan@itg.be A quick aside on who I am Tangents already! Who I am A postdoc at the Institute of Tropical Medicine in Antwerp, Belgium Mycobacteria evolution
Microbial analysis with STAMP Conor Meehan cmeehan@itg.be A quick aside on who I am Tangents already! Who I am A postdoc at the Institute of Tropical Medicine in Antwerp, Belgium Mycobacteria evolution
Phylogenetic inference
 Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Mitochondrial Genome Annotation
 Protein Genes 1,2 1 Institute of Bioinformatics University of Leipzig 2 Department of Bioinformatics Lebanese University TBI Bled 2015 Outline Introduction Mitochondrial DNA Problem Tools Training Annotation
Protein Genes 1,2 1 Institute of Bioinformatics University of Leipzig 2 Department of Bioinformatics Lebanese University TBI Bled 2015 Outline Introduction Mitochondrial DNA Problem Tools Training Annotation
An Introduction to Bioinformatics Algorithms Hidden Markov Models
 Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Pairwise sequence alignment and pair hidden Markov models
 Pairwise sequence alignment and pair hidden Markov models Martin C. Frith April 13, 2012 ntroduction Pairwise alignment and pair hidden Markov models (phmms) are basic textbook fare [2]. However, there
Pairwise sequence alignment and pair hidden Markov models Martin C. Frith April 13, 2012 ntroduction Pairwise alignment and pair hidden Markov models (phmms) are basic textbook fare [2]. However, there
Taxonomical Classification using:
 Taxonomical Classification using: Extracting ecological signal from noise: introduction to tools for the analysis of NGS data from microbial communities Bergen, April 19-20 2012 INTRODUCTION Taxonomical
Taxonomical Classification using: Extracting ecological signal from noise: introduction to tools for the analysis of NGS data from microbial communities Bergen, April 19-20 2012 INTRODUCTION Taxonomical
Hands-On Nine The PAX6 Gene and Protein
 Hands-On Nine The PAX6 Gene and Protein Main Purpose of Hands-On Activity: Using bioinformatics tools to examine the sequences, homology, and disease relevance of the Pax6: a master gene of eye formation.
Hands-On Nine The PAX6 Gene and Protein Main Purpose of Hands-On Activity: Using bioinformatics tools to examine the sequences, homology, and disease relevance of the Pax6: a master gene of eye formation.
A phylogenomic toolbox for assembling the tree of life
 A phylogenomic toolbox for assembling the tree of life or, The Phylota Project (http://www.phylota.org) UC Davis Mike Sanderson Amy Driskell U Pennsylvania Junhyong Kim Iowa State Oliver Eulenstein David
A phylogenomic toolbox for assembling the tree of life or, The Phylota Project (http://www.phylota.org) UC Davis Mike Sanderson Amy Driskell U Pennsylvania Junhyong Kim Iowa State Oliver Eulenstein David
MULTIPLE SEQUENCE ALIGNMENT FOR CONSTRUCTION OF PHYLOGENETIC TREE
 MULTIPLE SEQUENCE ALIGNMENT FOR CONSTRUCTION OF PHYLOGENETIC TREE Manmeet Kaur 1, Navneet Kaur Bawa 2 1 M-tech research scholar (CSE Dept) ACET, Manawala,Asr 2 Associate Professor (CSE Dept) ACET, Manawala,Asr
MULTIPLE SEQUENCE ALIGNMENT FOR CONSTRUCTION OF PHYLOGENETIC TREE Manmeet Kaur 1, Navneet Kaur Bawa 2 1 M-tech research scholar (CSE Dept) ACET, Manawala,Asr 2 Associate Professor (CSE Dept) ACET, Manawala,Asr
Investigation 3: Comparing DNA Sequences to Understand Evolutionary Relationships with BLAST
 Investigation 3: Comparing DNA Sequences to Understand Evolutionary Relationships with BLAST Introduction Bioinformatics is a powerful tool which can be used to determine evolutionary relationships and
Investigation 3: Comparing DNA Sequences to Understand Evolutionary Relationships with BLAST Introduction Bioinformatics is a powerful tool which can be used to determine evolutionary relationships and
Sequence Analysis, '18 -- lecture 9. Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene.
 Sequence Analysis, '18 -- lecture 9 Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene. How can I represent thousands of homolog sequences in a compact
Sequence Analysis, '18 -- lecture 9 Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene. How can I represent thousands of homolog sequences in a compact
Hidden Markov Models in computational biology. Ron Elber Computer Science Cornell
 Hidden Markov Models in computational biology Ron Elber Computer Science Cornell 1 Or: how to fish homolog sequences from a database Many sequences in database RPOBESEQ Partitioned data base 2 An accessible
Hidden Markov Models in computational biology Ron Elber Computer Science Cornell 1 Or: how to fish homolog sequences from a database Many sequences in database RPOBESEQ Partitioned data base 2 An accessible
Alignment principles and homology searching using (PSI-)BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)
BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)") Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Hidden Markov Models. Main source: Durbin et al., Biological Sequence Alignment (Cambridge, 98)
") Hidden Markov Models Main source: Durbin et al., Biological Sequence Alignment (Cambridge, 98) 1 The occasionally dishonest casino A P A (1) = P A (2) = = 1/6 P A->B = P B->A = 1/10 B P B (1)=0.1... P
Hidden Markov Models Main source: Durbin et al., Biological Sequence Alignment (Cambridge, 98) 1 The occasionally dishonest casino A P A (1) = P A (2) = = 1/6 P A->B = P B->A = 1/10 B P B (1)=0.1... P
Multiple Sequence Alignment: HMMs and Other Approaches
 Multiple Sequence Alignment: HMMs and Other Approaches Background Readings: Durbin et. al. Section 3.1, Ewens and Grant, Ch4. Wing-Kin Sung, Ch 6 Beerenwinkel N, Siebourg J. Statistics, probability, and
Multiple Sequence Alignment: HMMs and Other Approaches Background Readings: Durbin et. al. Section 3.1, Ewens and Grant, Ch4. Wing-Kin Sung, Ch 6 Beerenwinkel N, Siebourg J. Statistics, probability, and
Phylogenetic Networks, Trees, and Clusters
 Phylogenetic Networks, Trees, and Clusters Luay Nakhleh 1 and Li-San Wang 2 1 Department of Computer Science Rice University Houston, TX 77005, USA nakhleh@cs.rice.edu 2 Department of Biology University
Phylogenetic Networks, Trees, and Clusters Luay Nakhleh 1 and Li-San Wang 2 1 Department of Computer Science Rice University Houston, TX 77005, USA nakhleh@cs.rice.edu 2 Department of Biology University
Comparison of Three Fugal ITS Reference Sets. Qiong Wang and Jim R. Cole
 RDP TECHNICAL REPORT Created 04/12/2014, Updated 08/08/2014 Summary Comparison of Three Fugal ITS Reference Sets Qiong Wang and Jim R. Cole wangqion@msu.edu, colej@msu.edu In this report, we evaluate the
RDP TECHNICAL REPORT Created 04/12/2014, Updated 08/08/2014 Summary Comparison of Three Fugal ITS Reference Sets Qiong Wang and Jim R. Cole wangqion@msu.edu, colej@msu.edu In this report, we evaluate the
Overview of Research at Bioinformatics Lab
 Overview of Research at Bioinformatics Lab Li Liao Develop new algorithms and (statistical) learning methods that help solve biological problems > Capable of incorporating domain knowledge > Effective,
Overview of Research at Bioinformatics Lab Li Liao Develop new algorithms and (statistical) learning methods that help solve biological problems > Capable of incorporating domain knowledge > Effective,
394C, October 2, Topics: Mul9ple Sequence Alignment Es9ma9ng Species Trees from Gene Trees
 394C, October 2, 2013 Topics: Mul9ple Sequence Alignment Es9ma9ng Species Trees from Gene Trees Mul9ple Sequence Alignment Mul9ple Sequence Alignments and Evolu9onary Histories (the meaning of homologous
394C, October 2, 2013 Topics: Mul9ple Sequence Alignment Es9ma9ng Species Trees from Gene Trees Mul9ple Sequence Alignment Mul9ple Sequence Alignments and Evolu9onary Histories (the meaning of homologous
Basic Local Alignment Search Tool
 Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment
 Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Session 5: Phylogenomics
 Session 5: Phylogenomics B.- Phylogeny based orthology assignment REMINDER: Gene tree reconstruction is divided in three steps: homology search, multiple sequence alignment and model selection plus tree
Session 5: Phylogenomics B.- Phylogeny based orthology assignment REMINDER: Gene tree reconstruction is divided in three steps: homology search, multiple sequence alignment and model selection plus tree
Hidden Markov Models and Their Applications in Biological Sequence Analysis
 Hidden Markov Models and Their Applications in Biological Sequence Analysis Byung-Jun Yoon Dept. of Electrical & Computer Engineering Texas A&M University, College Station, TX 77843-3128, USA Abstract
Hidden Markov Models and Their Applications in Biological Sequence Analysis Byung-Jun Yoon Dept. of Electrical & Computer Engineering Texas A&M University, College Station, TX 77843-3128, USA Abstract
Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction. Lesser Tenrec (Echinops telfairi)
") Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction Lesser Tenrec (Echinops telfairi) Goals: 1. Use phylogenetic experimental design theory to select optimal taxa to
Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction Lesser Tenrec (Echinops telfairi) Goals: 1. Use phylogenetic experimental design theory to select optimal taxa to
HMM applications. Applications of HMMs. Gene finding with HMMs. Using the gene finder
 HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
Whole Genome Alignments and Synteny Maps
 Whole Genome Alignments and Synteny Maps IINTRODUCTION It was not until closely related organism genomes have been sequenced that people start to think about aligning genomes and chromosomes instead of
Whole Genome Alignments and Synteny Maps IINTRODUCTION It was not until closely related organism genomes have been sequenced that people start to think about aligning genomes and chromosomes instead of
HMMs and biological sequence analysis
 HMMs and biological sequence analysis Hidden Markov Model A Markov chain is a sequence of random variables X 1, X 2, X 3,... That has the property that the value of the current state depends only on the
HMMs and biological sequence analysis Hidden Markov Model A Markov chain is a sequence of random variables X 1, X 2, X 3,... That has the property that the value of the current state depends only on the
Bioinformatics. Scoring Matrices. David Gilbert Bioinformatics Research Centre
 Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Probalign: Multiple sequence alignment using partition function posterior probabilities
 Sequence Analysis Probalign: Multiple sequence alignment using partition function posterior probabilities Usman Roshan 1* and Dennis R. Livesay 2 1 Department of Computer Science, New Jersey Institute
Sequence Analysis Probalign: Multiple sequence alignment using partition function posterior probabilities Usman Roshan 1* and Dennis R. Livesay 2 1 Department of Computer Science, New Jersey Institute
How to read and make phylogenetic trees Zuzana Starostová
 How to read and make phylogenetic trees Zuzana Starostová How to make phylogenetic trees? Workflow: obtain DNA sequence quality check sequence alignment calculating genetic distances phylogeny estimation
How to read and make phylogenetic trees Zuzana Starostová How to make phylogenetic trees? Workflow: obtain DNA sequence quality check sequence alignment calculating genetic distances phylogeny estimation
Supplementary Information
 Supplementary Information Supplementary Figure 1. Schematic pipeline for single-cell genome assembly, cleaning and annotation. a. The assembly process was optimized to account for multiple cells putatively
Supplementary Information Supplementary Figure 1. Schematic pipeline for single-cell genome assembly, cleaning and annotation. a. The assembly process was optimized to account for multiple cells putatively
Phylogenetics in the Age of Genomics: Prospects and Challenges
 Phylogenetics in the Age of Genomics: Prospects and Challenges Antonis Rokas Department of Biological Sciences, Vanderbilt University http://as.vanderbilt.edu/rokaslab http://pubmed2wordle.appspot.com/
Phylogenetics in the Age of Genomics: Prospects and Challenges Antonis Rokas Department of Biological Sciences, Vanderbilt University http://as.vanderbilt.edu/rokaslab http://pubmed2wordle.appspot.com/
Phylogeny and systematics. Why are these disciplines important in evolutionary biology and how are they related to each other?
 Phylogeny and systematics Why are these disciplines important in evolutionary biology and how are they related to each other? Phylogeny and systematics Phylogeny: the evolutionary history of a species
Phylogeny and systematics Why are these disciplines important in evolutionary biology and how are they related to each other? Phylogeny and systematics Phylogeny: the evolutionary history of a species
Hidden Markov Models
 Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Evaluating the usefulness of alignment filtering methods to reduce the impact of errors on evolutionary inferences
 Di Franco et al. BMC Evolutionary Biology (2019) 19:21 https://doi.org/10.1186/s12862-019-1350-2 RESEARCH ARTICLE Evaluating the usefulness of alignment filtering methods to reduce the impact of errors
Di Franco et al. BMC Evolutionary Biology (2019) 19:21 https://doi.org/10.1186/s12862-019-1350-2 RESEARCH ARTICLE Evaluating the usefulness of alignment filtering methods to reduce the impact of errors
Phylogenetic Tree Reconstruction
 I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
17 Non-collinear alignment Motivation A B C A B C A B C A B C D A C. This exposition is based on:
 17 Non-collinear alignment This exposition is based on: 1. Darling, A.E., Mau, B., Perna, N.T. (2010) progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5(6):e11147.
17 Non-collinear alignment This exposition is based on: 1. Darling, A.E., Mau, B., Perna, N.T. (2010) progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5(6):e11147.
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Comparison of Cost Functions in Sequence Alignment. Ryan Healey
 Comparison of Cost Functions in Sequence Alignment Ryan Healey Use of Cost Functions Used to score and align sequences Mathematically model how sequences mutate and evolve. Evolution and mutation can be
Comparison of Cost Functions in Sequence Alignment Ryan Healey Use of Cost Functions Used to score and align sequences Mathematically model how sequences mutate and evolve. Evolution and mutation can be
Bioinformatics. Proteins II. - Pattern, Profile, & Structure Database Searching. Robert Latek, Ph.D. Bioinformatics, Biocomputing
 Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
8/23/2014. Phylogeny and the Tree of Life
 Phylogeny and the Tree of Life Chapter 26 Objectives Explain the following characteristics of the Linnaean system of classification: a. binomial nomenclature b. hierarchical classification List the major
Phylogeny and the Tree of Life Chapter 26 Objectives Explain the following characteristics of the Linnaean system of classification: a. binomial nomenclature b. hierarchical classification List the major
Motifs, Profiles and Domains. Michael Tress Protein Design Group Centro Nacional de Biotecnología, CSIC
 Motifs, Profiles and Domains Michael Tress Protein Design Group Centro Nacional de Biotecnología, CSIC Comparing Two Proteins Sequence Alignment Determining the pattern of evolution and identifying conserved
Motifs, Profiles and Domains Michael Tress Protein Design Group Centro Nacional de Biotecnología, CSIC Comparing Two Proteins Sequence Alignment Determining the pattern of evolution and identifying conserved
Page 1. References. Hidden Markov models and multiple sequence alignment. Markov chains. Probability review. Example. Markovian sequence
 Page Hidden Markov models and multiple sequence alignment Russ B Altman BMI 4 CS 74 Some slides borrowed from Scott C Schmidler (BMI graduate student) References Bioinformatics Classic: Krogh et al (994)
Page Hidden Markov models and multiple sequence alignment Russ B Altman BMI 4 CS 74 Some slides borrowed from Scott C Schmidler (BMI graduate student) References Bioinformatics Classic: Krogh et al (994)
Using phylogenetics to estimate species divergence times... Basics and basic issues for Bayesian inference of divergence times (plus some digression)
") Using phylogenetics to estimate species divergence times... More accurately... Basics and basic issues for Bayesian inference of divergence times (plus some digression) "A comparison of the structures
Using phylogenetics to estimate species divergence times... More accurately... Basics and basic issues for Bayesian inference of divergence times (plus some digression) "A comparison of the structures
Model Accuracy Measures
 Model Accuracy Measures Master in Bioinformatics UPF 2017-2018 Eduardo Eyras Computational Genomics Pompeu Fabra University - ICREA Barcelona, Spain Variables What we can measure (attributes) Hypotheses
Model Accuracy Measures Master in Bioinformatics UPF 2017-2018 Eduardo Eyras Computational Genomics Pompeu Fabra University - ICREA Barcelona, Spain Variables What we can measure (attributes) Hypotheses
Markov Models & DNA Sequence Evolution
 7.91 / 7.36 / BE.490 Lecture #5 Mar. 9, 2004 Markov Models & DNA Sequence Evolution Chris Burge Review of Markov & HMM Models for DNA Markov Models for splice sites Hidden Markov Models - looking under
7.91 / 7.36 / BE.490 Lecture #5 Mar. 9, 2004 Markov Models & DNA Sequence Evolution Chris Burge Review of Markov & HMM Models for DNA Markov Models for splice sites Hidden Markov Models - looking under
CHAPTERS 24-25: Evidence for Evolution and Phylogeny
 CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
Hidden Markov Models. based on chapters from the book Durbin, Eddy, Krogh and Mitchison Biological Sequence Analysis via Shamir s lecture notes
 Hidden Markov Models based on chapters from the book Durbin, Eddy, Krogh and Mitchison Biological Sequence Analysis via Shamir s lecture notes music recognition deal with variations in - actual sound -
Hidden Markov Models based on chapters from the book Durbin, Eddy, Krogh and Mitchison Biological Sequence Analysis via Shamir s lecture notes music recognition deal with variations in - actual sound -
BLAST Database Searching. BME 110: CompBio Tools Todd Lowe April 8, 2010
 BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for