Multiple sequence alignments
|
|
|
- Bethany Osborne
- 5 years ago
- Views:
Transcription




1 Multiple sequence alignments Special thanks to all the scientis that made public available their presentations throughout the web from where many slides were taken to eleborate this presentation Web sites used in our practice Figures are linked to their corresponding web sites What is a Multiple Sequence Alignment? Sequence Retrieval system RSA Tools ClustalW BLAST Structural Criteria Residues are arranged so that those playing a similar role end up in the same column. Evolutive Criteria Residues are arranged so that those having the same ancestor end up in the same column. Similarity Criteria As many similar residues as possible in the same column Enrique Merino, IBT-UNAM 1
2 2,000,000,000 years Alineamientos múltiples de secuencias Multiple sequence alignments Multiple sequence alignments Seems a simple extension: Align k sequences at the same time. AGTGACCTGGGAAGACCCTGACCCTGGGTCACAAAACTC AGTGCCCTGGAACCCTGACGGTGGGTCACAAAACTTCTGGA y z x Unfortunately, this can get very expensive. For more than eight proteins of average length, the problem is non-computable given current computer power. Therefore, all of the methods capable of handling larger problems in practical timescales make use of heuristics. Aligning N sequences of length L requires a matrix of size L N, where each square in the matrix has 2 N -1 neighbors This gives a total time complexity of O(2 N L N ) What is a Multiple Sequence Alignment? The MSA contains what you put inside You can view your MSA as: A record of evolution A summary of a protein family A collection of experiments made for you by Nature a MSA is a MODEL Enrique Merino, IBT-UNAM 2


3 What Is A Multiple Sequence Alignment? Why Is It Difficult To Compute A multiple Sequence Alignment? It Indicates the RELATIONSHIP between residues of different sequences. It REVEALS -Similarities -Inconsistencies Multiple Alignments are CENTRAL to MOST Bioinformatics Techniques. A CROSSROAD PROBLEM BIOLOGY: What is A Good Alignment COMPUTATION What is THE Good Alignment chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKD wheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSE trybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGP mouse -----KPKRPRSAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. :::.:... :.. *. *: * Why Is It Difficult To Compute A multiple Sequence Alignment? Multiple Alignments: What Are They Good For? BIOLOGY COMPUTATION CIRCULAR PROBLEM... Good Sequences Good Alignment Enrique Merino, IBT-UNAM 3
4 Multiple Sequence Alignment Derived Information motif Frequency (identity) matrices fingerprint Profile(gapped weight matrix) Multiple Sequence Alignment clasification Simultaneous As opposed to [Simultaneous: they simultaneously use all the information] Exact As opposed to Heursistic [Heuristics: cut corners like Blast Vs SW] [Heuristics: do not guarranty an optimal solution] RWDAGCVN RWDSGCVN RWHHGCVQ RWKGACYN RWLWACEQ Position-specific weight matrices (blocks) Hidden Markov model (HMM) Stochastic As opposed to Determinist [Stochastic: contain an element of randomness] [Stochastic: Example of a Monte Carlo Surface estimation ] Iterative As opposed to Non Iterative R-W-x(2)-[AG]-C-x-[NQ] Regular expression (pattern) [Iterative: run the same algorithm many times] [Iterative: Most stochastic methods are iterative] Exhaustive methods Heuristic methods The Correct Alignment Correct according to optimality criteria Always Not always Correct according to homology Not always Not always GAs Simultaneous MSA POA HMMs Iteralign Prrp DCA Combalign OMA GA SAM HMMer Iterative Clustal T-Coffee Praline MAFFT SAGA Dialign Non tree based Enrique Merino, IBT-UNAM 4

5 Simultaneous DCA Clustal MSA Combalign T-Coffee In any case, MSA consider the evolution of each column as independent process POA Iteralign Prrp OMA GA SAM HMMer Iterative Praline MAFFT SAGA Dialign Stochastic How close to reality is this assumption? 3D protein models can be evaluated based on the co-evolution of their interacting residues A B The presence of 'correlated positions' between pairs of positions in pairs of multiple sequence alignments can be used in predicting intra-protein and proteinprotein interactions. A B A B A B A B Enrique Merino, IBT-UNAM 5


6 Multiple sequence alignments. Clustal W Step 1 Pairwise Alignment. Compare each sequence with each other calculate a distance matrix human EYSGSSEKIDLLASDPHEALICKSERVHSKSVESNIEDKIFGKTYRKKASLPNLSHVTEN 480 Dog EYSGSSEKIDLMASDPQDAFICESERVHTKPVGGNIEDKIFGKTYRRKASLPKVSHTTEV 477 mouse GGFSSSRKTDLVTPDPHHTLMCKSGRDFSKPVEDNISDKIFGKSYQRKGSRPHLNHVTE 476 Julie D.Thompson, Desmond G.Higgins and Toby J.Gibson. Nucleic Acids Research, 1994, Vol. 22, No SeqA Name Len(aa) SeqB Name Len(aa) Identity 1 human 60 2 dog 60 77% 1 human 60 3 mouse 59 61% 2 dog 60 3 mouse 59 52% Compare each sequence with each other calculate a distance matrix Multiple sequence alignments. Clustal W Step 1 Pairwise Alignment. Compare each sequence with each other calculate a distance matrix Different sequences H D M H D M Distance = Number of exact matches divided by the sequence length (ignoring gaps). Thus, the higher the number the more closely related the two sequences are. In this distance matrix, the sequence of Human is 76% identical to the sequence of Dog Enrique Merino, IBT-UNAM 6
7 Step 2 Create Guide Tree. Use the results of the distance matrix to create a Guide Tree to help determine in what order the sequences will be aligned. H D M Multiple sequence alignments. Clustal W Initially the guide Trees were calculated using the UPGMA method. The current version uses the Neighbour-Joining method which gives better estimates of individual branch lengths H D M Guide Tree, or Dendrogram has no phylogenetic meaning Cannot be used to show evolutionary relationships H D M Guide Tree Step 3 Alignment Follow the Guide Tree and align the sequences A B C Multiple sequence alignments. Clustal W 1. Align Human and Dog first 2. Add sequence Mouse to the previous alignment of Human and Dog Align the most closely related sequences first, then add in the more distantly related ones and align them to the existing alignment, inserting gaps if necessary Multiple sequence alignments. Clustal W By the time the most distantly related sequences are aligned, one already has a sample of aligned sequences which gives important information about the variability at each position Multiple sequence alignments. Clustal W Gap treatment Short stretches of 5 hydrophilic residues often indicate loop or random coil regions (not essential for structure) and therefore gap penalties are reduced for such stretches. Gap penalties for closely related sequences are lowered compared to more distantly related sequences ( once a gap always a gap rule). It is thought that those gaps occur in regions that do not disrupt the structure or function. Alignments of proteins of known structure show that proteins gaps do not occur more frequently than every eight residues. Therefore penalties for gaps increase when required at 8 residues or less for alignment. This gives a lower alignment score in that region. A gap weight is assigned after each aa according the frequency that such a gap naturally occurs after that aa in nature Enrique Merino, IBT-UNAM 7

8 Multiple sequence alignments. Clustal W Amino acid weight matrices As we know, there are many scoring matrices that one can use depending on the relatedness of the aligned proteins. As the alignment proceeds to longer branches the aa scoring matrices are changed to accommodate more divergent sequences. The length of the branch is used to determine which matrix to use. Similar sequences with "hard" matrices (BLOSUM80) Distant sequences with "soft" matrices (BLOSUM50) Multiple sequence alignments. Clustal W Relative contribution of each pairwise alignment to the global alignment score Sequences are weighted to compensate for bias of redundant elements in the alignment Flowchart of computation steps in Clustal Pairwise Alignment: Calculation of distance matrix Creation of unrooted Neighbor-Joining Tree Rooted NJ Tree (guide tree) and calculation of sequence weights alignment following the Guide Tree Enrique Merino, IBT-UNAM 8




9 ClustalW Multiple sequence alignments. TCoffee Multiple sequence alignments. TCoffee T-Coffee: Mixing Local and Global Alignments Regular progressive alignment strategy may produce alignment errors Local Alignment Global Alignment The global alignments are constructed using ClustalW on the sequences, two at a time Extension Library Based Multiple Sequence Alignment Multiple Sequence Alignment The local alignments are the ten top scoring nonintersecting local alignments, between each pair of sequences, gathered using the Lalign program (which is a variant of the Smith and Waterman Method) of the FASTA package Enrique Merino, IBT-UNAM 9


10 T-Coffee: Primary Library T-Coffee: Analysis of Consistency In the library, each alignment is represented as a list of pair-wise residue matches, each of these pairs is a constraint. All of these constraints are not equally important. This data is taken into account when computing the multiple alignment and give priority to the most reliable residue pairs We enormously increase the value of the information in the library by examining the consistency of each pair of residues with residue pairs from all of the other alignments. For each pair of aligned residues in the library, we can assign a weight that reflects the degree to which those residues align consistently with residues The Triplet Assumption SEQ A Y Y SEQ B Z Consistency Consensus ClustalW T-Coffee Enrique Merino, IBT-UNAM 10
 Multiple Alignment You can t satisfy them all You must satisfy as many as possible (The easy ones) Specialist Structural Multiple Sequence")
11 Alignment T-Coffee: Alignmed sequences using Extend Library Dynamic Programming Using An Extended Library T-Coffee and Concistency Mixing Heterogenous Data With T-Coffee Local Alignment Global Alignment Each Library Line is a Soft Constraint (a wish) Multiple Alignment You can t satisfy them all You must satisfy as many as possible (The easy ones) Specialist Structural Multiple Sequence Alignment Enrique Merino, IBT-UNAM 11

12 T-Coffee and Consistency (Summary) The method is broadly based on the popular progressive approach to multiple alignment but avoids the most serious pitfalls caused by the greedy nature of this algorithm. With T-Coffee we pre-process a data set of all pair-wise alignments between the sequences. 3D-Coffee Why Do We Want To Mix Sequences and Structures? This provides us with a library of alignment information that can be used to guide the progressive alignment. Intermediate alignments are then based not only on the sequences to be aligned next but also on how all of the sequences align with each other. This alignment information can be derived from heterogeneous sources such as a mixture of alignment programs and/or structure superposition. Sequences are Cheap and Common. Structures are Expensive and Rare. 3D-Coffee Why Do We Want To Mix Sequences and Structures? Cheapest Structure determination: Sequence-Structure Alignment Convincing Alignment Same Fold 3D-Coffee Why Do We Want To Mix Sequences and Structures? Distant sequences are hard to align THREAD Or ALIGN ADKPRRP---LS-YMLWLN ADKPKRPKPRLSAYMLWLN Enrique Merino, IBT-UNAM 12


13 3D-Coffee Why Do We Want To Mix Sequences and Structures? 3D-Coffee Why Do We Want To Mix Sequences and Structures? chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKD wheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSE trybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGP mouse -----KPKRPRSAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. :::.:... :.. *. *: * Multiple Sequence Alignments Help Exploring the Twilight Zone Structure Superposition 3D-Coffee Why Do We Want To Mix Sequences and Structures? Conclusion -Structures Help BUT NOT SO MUCH Enrique Merino, IBT-UNAM 13



14 MUSCLE Multiple Sequence Alignment with reduced time and space complexity MUSCLE Multiple Sequence Alignment with reduced time and space complexity Basic Strategy: A progressive alignment is built, to which horizontal refinement is applied Three stages At end of each stage, a multiple alignment is available and the algorithm can be terminated MUSCLE Multiple Sequence Alignment with reduced time and space complexity Draft Improved Refinement Enrique Merino, IBT-UNAM 14
15 MUSCLE. Stage 1. Draft 1.1. Similarity measure and Distance estimate Draft Improved MUSCLE. Stage 1. Draft 1.1. Similarity measure and Distance estimate The goal of the first stage is to produce a multiple alignment, emphasizing speed over accuracy Calculated using k-mer counting. A kmer is a contiguous subsequence of length k, also known as a word or k-tuple. Related sequences tend to have more kmers in common than expected by chance Refinement k-mer: ATG CCA ACCATGCGAATGGTCCACAATG score: 3 2 MUSCLE. Stage 1. Draft 1.1. Similarity measure and Distance estimate Based on the pairwise similarities, a triangular distance matrix is computed. MUSCLE. Stage 1. Draft 1.2. Tree construction using UPGMA Draft Improved Refinement Enrique Merino, IBT-UNAM 15

16 MUSCLE. Stage 1. Draft 1.2. Tree construction using UPGMA From the distance matrix we construct a tree using the UPGM method MUSCLE. Stage 1. Draft 1.2. Tree construction using UPGMA (Unweighted Pair Group Method with Arithmetic mean) One of the fastest and tree construction methods Is a simple agglomerative or bottom-up data clustering method UPGMA assumes a constant rate of evolution (molecular clock hypothesis). At each step, the nearest 2 clusters are combined into a higher-level cluster. The distance between any 2 clusters A and B is taken to be the average of all distances between pairs of objects "a" in A and "b" in B. MUSCLE. Stage 1. Draft MUSCLE. Stage 1. Draft 1.3. alignment. Draft Improved 1.3. alignment. A progressive alignment is built by following the branching order of the tree, yielding a multiple alignment of all input sequences at the root. The alignment is done by profiles Profile-profile alignment Refinement Enrique Merino, IBT-UNAM 16
17 MUSCLE. Stage 1. Draft 1.3. alignment. A progressive alignment is built by following the branching order of the tree, yielding a multiple alignment of all input sequences at the root. The alignment is done by profiles alignment MUSCLE. Stage 2. Improved 2.1. Similarity measure and Distance estimate Draft Improved Refinement MUSCLE. Stage 2. Improved 2.1. Similarity measure and Distance estimate MUSCLE. Stage 2. Improved 2.1. Similarity measure and Distance estimate The main source of error in the draft progressive stage is the approximate kmer distance measure, which results in a suboptimal tree. MUSCLE therefore re-estimates the tree using the Kimura distance, which is more accurate but requires an alignment Enrique Merino, IBT-UNAM 17
18 MUSCLE. Stage 2. Improved 2.2. Tree construction using UPGMA Draft MUSCLE. Stage 2. Improved 2.2. Tree construction using UPGMA A tree is constructed by computing a Kimura distance matrix and applying a clustering method to it Improved Refinement 3 4 MUSCLE. Stage 2. Improved 2.3. alignment Draft MUSCLE. Stage 2. Improved 2.3. alignment A new progressive alignment is built Improved Refinement New Alignment Enrique Merino, IBT-UNAM 18
19 MUSCLE. Stage 2. Improved 2.4. Tree comparison The new tree is compared to the previous tree by identifying the set of internal nodes for which the branching order has changed. If Stage 2 has executed more than once, and the number of changed nodes has not decreased, the process of improving the tree is considered to have converged and iteration terminates. MUSCLE. Stage 3. Refinement. Refinement is performed iteratively MUSCLE. Stage 3. Refinement Delete edge from the Tree. Draft MUSCLE. Stage 3. Refinement Delete edge from the Tree. Choice of bipartition An edge is removed from the tree, dividing the sequences into two disjoint subsets Improved Refinement Enrique Merino, IBT-UNAM 19
20 MUSCLE. Stage 3. Refinement Compute subtree profiles. MUSCLE. Stage 3. Refinement Compute subtree profiles. Draft The multiple alignment of each subset is extracted from current multiple alignment. Columns made up of indels only are removed Improved Refinement TCC--AA TCA--GA TCA--AA G--ATAC T--CTGC TCC--AA TCA--AA TCA--GA G--ATAC T--CTGC TCCAA TCAAA MUSCLE. Stage 3. Refinement Re-align profiles. MUSCLE. Stage 3. Refinement Re-align profiles. Draft The two profiles are then realigned with each other using profile-profile alignment. Improved Refinement TCCAA TCAAA TCA--GA G--ATAC T--CTGC T--CCAA T--CAAA TCA--GA G--ATAC T--CTGC Enrique Merino, IBT-UNAM 20
21 MUSCLE. Stage 3. Refinement Accept/Reject. MUSCLE. Stage 3. Refinement Accept/Reject. Draft The score of the new alignment is computed, if the score is higher than the old alignment, the new alignment is retained, otherwise it is discarded. Improved Refinement New T--CCAA T--CAAA TCA--GA G--ATAC T--CTGC OR Old TCC--AA TCA--GA TCA--AA G--ATAC T--CTGC MUSCLE. Stage 3. Refinement Accept/Reject. MUSCLE Multiple Sequence Alignment with reduced time and space complexity 1234 Score of alignment ACGT match=1 ACGA mismatch=0 AGGA T-coffee 1: A-A + A-A + A-A = = 3 2: C-C + C-G + C-G =1+0+0 = 1 3: G-G + G-G + G-G = = 3 4: T-A + T-A + A-A = =1 MUSCLE S(alignment) = S(1) + S(2) + S(3) + S(4) = = 8 The higher the score, the better the alignment Enrique Merino, IBT-UNAM 21



 TRAP is form of 11 identical")
22 An incorrect conclusion may come from a sequence alignment using incorrect assumptions Identification of TRAP orthologs as an example of the risk of common mistakes in the analysis Supose you want to align a set of MtrB sequences retrived by gene name fromncbi Angela Valbuzzi and Charles Yanofsky. SCIENCE VOL SEPTEMBER 2001 Computatonal Genomic group Insted of Ribosome, attenuation is mediated by an RNA binding protein called TRAP (trp RNA-Binding Attenuation Protein ) In Bacillus subtilis the trp operon is also regulated by transcription attenuation Secuencias TRAP (TRyptophan Attenuation Protein) TRAP is form of 11 identical subunits Biología Computacional Biología Computacional Enrique Merino, IBT-UNAM 22




23 An incorrect conclusion may come from a sequence alignment using incorrect assumptions An incorrect conclusion may come from a sequence alignment using incorrect assumptions Supose you want to align a set of MtrB sequences retrived by gene name fromncbi MtrB [Desulfobacterium autotrophicum HRM2] Signal transduction histidine kinase, nitrate/nitrite-specific MtrB [Bacillus amyloliquefaciens FZB42] Tryptophan RNA-binding attenuator protein Never forget that MSA is just a model that performs on a set of sequences given by the user Enrique Merino, IBT-UNAM 23



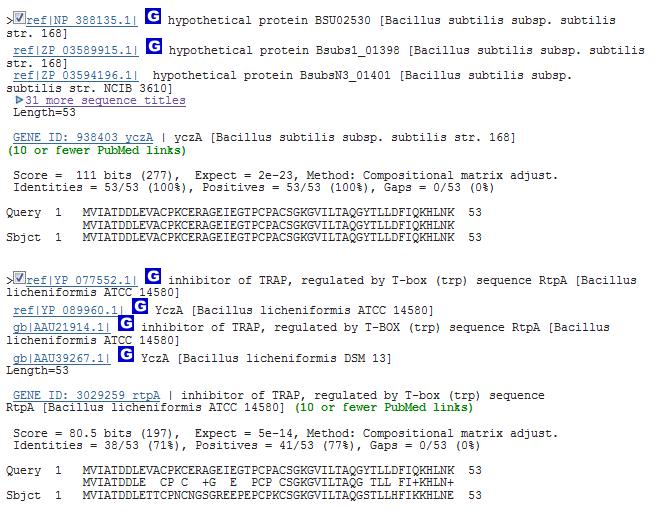
24 Exercise: Multiple sequence alignment Use multiple sequence alignment to analyze how our model antitrap align with their corresponding likely long distant homologs. Use multiple sequence alignment to analyze how antitrap align with their corresponding likely long distant homologs Sequence search based on antitrap Protein sequence Use multiple sequence alignment to analyze how antitrap align with their corresponding likely long distant homologs Use multiple sequence alignment to analyze how antitrap align with their corresponding likely long distant homologs Enrique Merino, IBT-UNAM 24

25 Use multiple sequence alignment to analyze how antitrap align with their corresponding likely long distant homologs >gi ref NP_ hypothetical protein BSU02530 [Bacillus subtilis subsp. subtilis str. 168] MVIATDDLEVACPKCERAGEIEGTPCPACSGKGVILTAQGYTLLDFIQKHLNK >gi ref YP_ inhibitor of TRAP, regulated by T-box (trp) sequence RtpA [Bacillus licheniformis ATCC 14580] MVIATDDLETTCPNCNGSGREEPEPCPKCSGKGVILTAQGSTLLHFIKKHLNE >gi ref YP_ RtpA [Bacillus amyloliquefaciens FZB42] MTGDGQTIKKGGIFMVIATDDLELTCPHCEGTGEEKEGTPCPKCGAKGVILTAQGNTLLHFIRKHIDQ >gi ref YP_ hypothetical protein Sfum_2476 [Syntrophobacter fumaroxidans MPOB] MVRMRLPELETKCWMCWGSGKIASEDHGGGMECPECGGVGWLPTADGRRLLDFVQRHLGIVEEGEDNETL >gi ref ZP_ chaperone protein DnaJ [Atopobium rimae ATCC 49626] MASMNEKDYYVILEVSETATTEEIRKAFQVKARKLHPDVNKAPDAEARFKEVSEAYAVLSDEGKRRRYDA MRSGNPFAGGYGPSGSPAGSNSYGQDPFGWGFPFGGVDFSSWRSQGSRRSRAYKPQTGADIEYDLTLTPM QAQEGVRKGITYQRFSACEACHGSGSVHHSEASSTCPTCGGTGHIHVDLSGIFGFGTVEMECPECEGTGH VVADPCEACGGSGRVLSASEAVVNVPPHAHDGDEIRMEGKGNAGTNGSKTGDFVVRVRVPEEQVTLRQSM GARAIGIALPFFAVDLATGASLLGTIIVAMLVVFGVRNIVGDGIKRSQRWWRNLGYAVVNGALTGIAWAL VAYMFFSCTAGLGRW >gi ref YP_ chaperone protein DnaJ [Nautilia profundicola AmH] MDYYEILGVERTATKVEIKKAYRKLAMKYHPDKNPGDKEAEEMFKKINEAYQVLSDDEKRAIYDKYGKEG LEGQGFKTDFDFGDIFDMFNDIFGGGFGGGRAEVQMPYDIDKAIEVTLEFEEAVYGVSKEIEINYFKLCP KCKGSGAEEKETCPSCHGRGTIIMGNGFMRISQTCPQCSGRGFIAKKVCNECRGKGYIVESETVKVDIPA GIDTGMRMRVKGRGNQDISGYRGDLYLIFNVKESKIFKRKGNNLIVEVPIFFTSAILGDTVKIPTLSGEK EIEIKPHTKDNTKIVFRGEGIADPNTGYRGDLIAILKIVYPKKLTDEQRELLEKLHKSFGGEIKEHKSIL EEAIDKVKSWFKGS >gi ref YP_ dnaj protein [Staphylococcus epidermidis RP62A] MAKRDYYEVLGVNKSASKDEIKKAYRKLSKKYHPDINKEEGADEKFKEISEAYEVLSDENKRVNYDQFGH DGPQGGFGSQGFGGSDFGGFEDIFSSFFGGGSRQRDPNAPRKGDDLQYTMTITFEEAVFGTKKEISIKKD VTCHTCNGDGAKPGTSKKTCSYCNGAGRVSVEQNTILGRVRTEQVCPKCEGSGQEFEEPCPTCKGKGTEN KTVKLEVTVPEGVDNEQQVRLAGEGSPGVNGGPHGDLYVVFRVKPSNTFERDGDDIYYNLDISFSQAALG DEIKIPTLKSNVVLTIPAGTQTGKQFRLKDKGVKNVHGYGYGDLFVNIKVVTPTKLNDRQKELLKEFAEI NGENINEQSSNFKDRAKRFFKGE Use multiple sequence alignment to analyze how antitrap align with their corresponding likely long distant homologs UPGMA UPGMA tree Unweighted Pair Group Method with Arithmetic mean One of the fastest and tree construction methods Used in Pileup (GCG package) Clustal uses neighbor joining, but calculating NJ tree is much more demanding; thus, UPGMA is demonstrated here Enrique Merino, IBT-UNAM 25
26 Constructing MSA human ACGTACGTCC gorilla ACCACCGTCC chimp ACCTACGTCC orangutan ACCCCCCTCC human ACGTACGTCC chimp ACCTACGTCC MUSCLE. Stage 2: Improved Similarity Measure Similarity is calculated for each pair of sequences using fractional identity computed from their mutual alignment in the current multiple alignment gorilla ACCACCGTCC orangutan ACCCCCCTCC human ACGTACGTCC chimp ACCTACGTCC gorilla ACCACCGTCC orangutan ACCCCCCTCC maqaque CCCCCCCCCC TCC--AA TCA--GA TCA--AA G--ATAC T--CTGC TCC--AA TCA--AA MUSCLE Multiple Sequence Alignment with reduced time and space complexity Enrique Merino, IBT-UNAM 26
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Multiple sequence alignment
 Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
CONCEPT OF SEQUENCE COMPARISON. Natapol Pornputtapong 18 January 2018
 CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
Copyright 2000 N. AYDIN. All rights reserved. 1
 Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT
 5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
Phylogenetic Tree Reconstruction
 I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
"Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky
 MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
Pairwise & Multiple sequence alignments
 Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
InDel 3-5. InDel 8-9. InDel 3-5. InDel 8-9. InDel InDel 8-9
 Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
Phylogenetic inference
 Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Multiple Sequence Alignment: A Critical Comparison of Four Popular Programs
 Multiple Sequence Alignment: A Critical Comparison of Four Popular Programs Shirley Sutton, Biochemistry 218 Final Project, March 14, 2008 Introduction For both the computational biologist and the research
Multiple Sequence Alignment: A Critical Comparison of Four Popular Programs Shirley Sutton, Biochemistry 218 Final Project, March 14, 2008 Introduction For both the computational biologist and the research
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Multiple Sequence Alignment. Sequences
 Multiple Sequence Alignment Sequences > YOR020c mstllksaksivplmdrvlvqrikaqaktasglylpe knveklnqaevvavgpgftdangnkvvpqvkvgdqvl ipqfggstiklgnddevilfrdaeilakiakd > crassa mattvrsvksliplldrvlvqrvkaeaktasgiflpe
Multiple Sequence Alignment Sequences > YOR020c mstllksaksivplmdrvlvqrikaqaktasglylpe knveklnqaevvavgpgftdangnkvvpqvkvgdqvl ipqfggstiklgnddevilfrdaeilakiakd > crassa mattvrsvksliplldrvlvqrvkaeaktasgiflpe
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT
 3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
Sequence Analysis '17- lecture 8. Multiple sequence alignment
 Sequence Analysis '17- lecture 8 Multiple sequence alignment Ex5 explanation How many random database search scores have e-values 10? (Answer: 10!) Why? e-value of x = m*p(s x), where m is the database
Sequence Analysis '17- lecture 8 Multiple sequence alignment Ex5 explanation How many random database search scores have e-values 10? (Answer: 10!) Why? e-value of x = m*p(s x), where m is the database
Sequence Analysis and Databases 2: Sequences and Multiple Alignments
 1 Sequence Analysis and Databases 2: Sequences and Multiple Alignments Jose María González-Izarzugaza Martínez CNIO Spanish National Cancer Research Centre (jmgonzalez@cnio.es) 2 Sequence Comparisons:
1 Sequence Analysis and Databases 2: Sequences and Multiple Alignments Jose María González-Izarzugaza Martínez CNIO Spanish National Cancer Research Centre (jmgonzalez@cnio.es) 2 Sequence Comparisons:
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Introduction to Bioinformatics Online Course: IBT
 Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
Single alignment: Substitution Matrix. 16 march 2017
 Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Ch. 9 Multiple Sequence Alignment (MSA)
") Ch. 9 Multiple Sequence Alignment (MSA) - gather seqs. to make MSA - doing MSA with ClustalW - doing MSA with Tcoffee - comparing seqs. that cannot align Introduction - from pairwise alignment to MSA -
Ch. 9 Multiple Sequence Alignment (MSA) - gather seqs. to make MSA - doing MSA with ClustalW - doing MSA with Tcoffee - comparing seqs. that cannot align Introduction - from pairwise alignment to MSA -
9/30/11. Evolution theory. Phylogenetic Tree Reconstruction. Phylogenetic trees (binary trees) Phylogeny (phylogenetic tree)
 Phylogeny (phylogenetic tree)") I9 Introduction to Bioinformatics, 0 Phylogenetic ree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & omputing, IUB Evolution theory Speciation Evolution of new organisms is driven by
I9 Introduction to Bioinformatics, 0 Phylogenetic ree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & omputing, IUB Evolution theory Speciation Evolution of new organisms is driven by
Sara C. Madeira. Universidade da Beira Interior. (Thanks to Ana Teresa Freitas, IST for useful resources on this subject)
") Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Effects of Gap Open and Gap Extension Penalties
 Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment
 Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
08/21/2017 BLAST. Multiple Sequence Alignments: Clustal Omega
 BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Multiple sequence alignment
 Multiple sequence alignment Wednesday, October 11, 2006 Sarah Wheelan swheelan@jhmi.edu Copyright notice Many of the images in this powerpoint presentation are from Bioinformatics and Functional Genomics
Multiple sequence alignment Wednesday, October 11, 2006 Sarah Wheelan swheelan@jhmi.edu Copyright notice Many of the images in this powerpoint presentation are from Bioinformatics and Functional Genomics
Genomics and bioinformatics summary. Finding genes -- computer searches
 Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Moreover, the circular logic
 Moreover, the circular logic How do we know what is the right distance without a good alignment? And how do we construct a good alignment without knowing what substitutions were made previously? ATGCGT--GCAAGT
Moreover, the circular logic How do we know what is the right distance without a good alignment? And how do we construct a good alignment without knowing what substitutions were made previously? ATGCGT--GCAAGT
MULTIPLE SEQUENCE ALIGNMENT FOR CONSTRUCTION OF PHYLOGENETIC TREE
 MULTIPLE SEQUENCE ALIGNMENT FOR CONSTRUCTION OF PHYLOGENETIC TREE Manmeet Kaur 1, Navneet Kaur Bawa 2 1 M-tech research scholar (CSE Dept) ACET, Manawala,Asr 2 Associate Professor (CSE Dept) ACET, Manawala,Asr
MULTIPLE SEQUENCE ALIGNMENT FOR CONSTRUCTION OF PHYLOGENETIC TREE Manmeet Kaur 1, Navneet Kaur Bawa 2 1 M-tech research scholar (CSE Dept) ACET, Manawala,Asr 2 Associate Professor (CSE Dept) ACET, Manawala,Asr
C3020 Molecular Evolution. Exercises #3: Phylogenetics
 C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
Similarity or Identity? When are molecules similar?
 Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Comparative Bioinformatics Midterm II Fall 2004
 Comparative Bioinformatics Midterm II Fall 2004 Objective Answer, part I: For each of the following, select the single best answer or completion of the phrase. (3 points each) 1. Deinococcus radiodurans
Comparative Bioinformatics Midterm II Fall 2004 Objective Answer, part I: For each of the following, select the single best answer or completion of the phrase. (3 points each) 1. Deinococcus radiodurans
Biochemistry 324 Bioinformatics. Pairwise sequence alignment
 Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Molecular Evolution and Phylogenetic Tree Reconstruction
 1 4 Molecular Evolution and Phylogenetic Tree Reconstruction 3 2 5 1 4 2 3 5 Orthology, Paralogy, Inparalogs, Outparalogs Phylogenetic Trees Nodes: species Edges: time of independent evolution Edge length
1 4 Molecular Evolution and Phylogenetic Tree Reconstruction 3 2 5 1 4 2 3 5 Orthology, Paralogy, Inparalogs, Outparalogs Phylogenetic Trees Nodes: species Edges: time of independent evolution Edge length
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5
 Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
Bioinformatics 1. Sepp Hochreiter. Biology, Sequences, Phylogenetics Part 4. Bioinformatics 1: Biology, Sequences, Phylogenetics
 Bioinformatics 1 Biology, Sequences, Phylogenetics Part 4 Sepp Hochreiter Klausur Mo. 30.01.2011 Zeit: 15:30 17:00 Raum: HS14 Anmeldung Kusss Contents Methods and Bootstrapping of Maximum Methods Methods
Bioinformatics 1 Biology, Sequences, Phylogenetics Part 4 Sepp Hochreiter Klausur Mo. 30.01.2011 Zeit: 15:30 17:00 Raum: HS14 Anmeldung Kusss Contents Methods and Bootstrapping of Maximum Methods Methods
Bioinformatics tools for phylogeny and visualization. Yanbin Yin
 Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
Phylogenies Scores for Exhaustive Maximum Likelihood and Parsimony Scores Searches
 Int. J. Bioinformatics Research and Applications, Vol. x, No. x, xxxx Phylogenies Scores for Exhaustive Maximum Likelihood and s Searches Hyrum D. Carroll, Perry G. Ridge, Mark J. Clement, Quinn O. Snell
Int. J. Bioinformatics Research and Applications, Vol. x, No. x, xxxx Phylogenies Scores for Exhaustive Maximum Likelihood and s Searches Hyrum D. Carroll, Perry G. Ridge, Mark J. Clement, Quinn O. Snell
Computational Biology
 Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
SUPPLEMENTARY INFORMATION
 Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
 Tree of Life iological Sequence nalysis Chapter http://tolweb.org/tree/ Phylogenetic Prediction ll organisms on Earth have a common ancestor. ll species are related. The relationship is called a phylogeny
Tree of Life iological Sequence nalysis Chapter http://tolweb.org/tree/ Phylogenetic Prediction ll organisms on Earth have a common ancestor. ll species are related. The relationship is called a phylogeny
Evolutionary Tree Analysis. Overview
 CSI/BINF 5330 Evolutionary Tree Analysis Young-Rae Cho Associate Professor Department of Computer Science Baylor University Overview Backgrounds Distance-Based Evolutionary Tree Reconstruction Character-Based
CSI/BINF 5330 Evolutionary Tree Analysis Young-Rae Cho Associate Professor Department of Computer Science Baylor University Overview Backgrounds Distance-Based Evolutionary Tree Reconstruction Character-Based
Multiple Sequence Alignment
 Multiple equence lignment Four ami Khuri Dept of omputer cience an José tate University Multiple equence lignment v Progressive lignment v Guide Tree v lustalw v Toffee v Muscle v MFFT * 20 * 0 * 60 *
Multiple equence lignment Four ami Khuri Dept of omputer cience an José tate University Multiple equence lignment v Progressive lignment v Guide Tree v lustalw v Toffee v Muscle v MFFT * 20 * 0 * 60 *
Phylogeny and Evolution. Gina Cannarozzi ETH Zurich Institute of Computational Science
 Phylogeny and Evolution Gina Cannarozzi ETH Zurich Institute of Computational Science History Aristotle (384-322 BC) classified animals. He found that dolphins do not belong to the fish but to the mammals.
Phylogeny and Evolution Gina Cannarozzi ETH Zurich Institute of Computational Science History Aristotle (384-322 BC) classified animals. He found that dolphins do not belong to the fish but to the mammals.
Inferring phylogeny. Constructing phylogenetic trees. Tõnu Margus. Bioinformatics MTAT
 Inferring phylogeny Constructing phylogenetic trees Tõnu Margus Contents What is phylogeny? How/why it is possible to infer it? Representing evolutionary relationships on trees What type questions questions
Inferring phylogeny Constructing phylogenetic trees Tõnu Margus Contents What is phylogeny? How/why it is possible to infer it? Representing evolutionary relationships on trees What type questions questions
Multiple sequence alignment
 Multiple sequence alignment Irit Orr Shifra Ben-Dor An example of Multiple Alignment VTISCTGSSSNIGAG-NHVKWYQQLPGQLPG VTISCTGTSSNIGS--ITVNWYQQLPGQLPG LRLSCSSSGFIFSS--YAMYWVRQAPG LSLTCTVSGTSFDD--YYSTWVRQPPG
Multiple sequence alignment Irit Orr Shifra Ben-Dor An example of Multiple Alignment VTISCTGSSSNIGAG-NHVKWYQQLPGQLPG VTISCTGTSSNIGS--ITVNWYQQLPGQLPG LRLSCSSSGFIFSS--YAMYWVRQAPG LSLTCTVSGTSFDD--YYSTWVRQPPG
In-Depth Assessment of Local Sequence Alignment
 2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
Study and Implementation of Various Techniques Involved in DNA and Protein Sequence Analysis
 Study and Implementation of Various Techniques Involved in DNA and Protein Sequence Analysis Kumud Joseph Kujur, Sumit Pal Singh, O.P. Vyas, Ruchir Bhatia, Varun Singh* Indian Institute of Information
Study and Implementation of Various Techniques Involved in DNA and Protein Sequence Analysis Kumud Joseph Kujur, Sumit Pal Singh, O.P. Vyas, Ruchir Bhatia, Varun Singh* Indian Institute of Information
Protein folding. α-helix. Lecture 21. An α-helix is a simple helix having on average 10 residues (3 turns of the helix)
") Computat onal Biology Lecture 21 Protein folding The goal is to determine the three-dimensional structure of a protein based on its amino acid sequence Assumption: amino acid sequence completely and uniquely
Computat onal Biology Lecture 21 Protein folding The goal is to determine the three-dimensional structure of a protein based on its amino acid sequence Assumption: amino acid sequence completely and uniquely
Motivating the need for optimal sequence alignments...
 1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
EVOLUTIONARY DISTANCES
 EVOLUTIONARY DISTANCES FROM STRINGS TO TREES Luca Bortolussi 1 1 Dipartimento di Matematica ed Informatica Università degli studi di Trieste luca@dmi.units.it Trieste, 14 th November 2007 OUTLINE 1 STRINGS:
EVOLUTIONARY DISTANCES FROM STRINGS TO TREES Luca Bortolussi 1 1 Dipartimento di Matematica ed Informatica Università degli studi di Trieste luca@dmi.units.it Trieste, 14 th November 2007 OUTLINE 1 STRINGS:
Comparative genomics: Overview & Tools + MUMmer algorithm
 Comparative genomics: Overview & Tools + MUMmer algorithm Urmila Kulkarni-Kale Bioinformatics Centre University of Pune, Pune 411 007. urmila@bioinfo.ernet.in Genome sequence: Fact file 1995: The first
Comparative genomics: Overview & Tools + MUMmer algorithm Urmila Kulkarni-Kale Bioinformatics Centre University of Pune, Pune 411 007. urmila@bioinfo.ernet.in Genome sequence: Fact file 1995: The first
Introduction to Evolutionary Concepts
 Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2009 VMD/MultiSeq
Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2009 VMD/MultiSeq
Sequence analysis and Genomics
 Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut
 Faculty of Veterinary Medicine Assiut") Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
Similarity searching summary (2)
") Similarity searching / sequence alignment summary Biol4230 Thurs, February 22, 2016 Bill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 What have we covered? Homology excess similiarity but no excess similarity
Similarity searching / sequence alignment summary Biol4230 Thurs, February 22, 2016 Bill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 What have we covered? Homology excess similiarity but no excess similarity
BINF6201/8201. Molecular phylogenetic methods
 BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
Whole Genome Alignments and Synteny Maps
 Whole Genome Alignments and Synteny Maps IINTRODUCTION It was not until closely related organism genomes have been sequenced that people start to think about aligning genomes and chromosomes instead of
Whole Genome Alignments and Synteny Maps IINTRODUCTION It was not until closely related organism genomes have been sequenced that people start to think about aligning genomes and chromosomes instead of
Phylogeny: building the tree of life
 Phylogeny: building the tree of life Dr. Fayyaz ul Amir Afsar Minhas Department of Computer and Information Sciences Pakistan Institute of Engineering & Applied Sciences PO Nilore, Islamabad, Pakistan
Phylogeny: building the tree of life Dr. Fayyaz ul Amir Afsar Minhas Department of Computer and Information Sciences Pakistan Institute of Engineering & Applied Sciences PO Nilore, Islamabad, Pakistan
A bioinformatics approach to the structural and functional analysis of the glycogen phosphorylase protein family
 A bioinformatics approach to the structural and functional analysis of the glycogen phosphorylase protein family Jieming Shen 1,2 and Hugh B. Nicholas, Jr. 3 1 Bioengineering and Bioinformatics Summer
A bioinformatics approach to the structural and functional analysis of the glycogen phosphorylase protein family Jieming Shen 1,2 and Hugh B. Nicholas, Jr. 3 1 Bioengineering and Bioinformatics Summer
Some Problems from Enzyme Families
 Some Problems from Enzyme Families Greg Butler Department of Computer Science Concordia University, Montreal www.cs.concordia.ca/~faculty/gregb gregb@cs.concordia.ca Abstract I will discuss some problems
Some Problems from Enzyme Families Greg Butler Department of Computer Science Concordia University, Montreal www.cs.concordia.ca/~faculty/gregb gregb@cs.concordia.ca Abstract I will discuss some problems
Alignment principles and homology searching using (PSI-)BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)
BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)") Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
How to read and make phylogenetic trees Zuzana Starostová
 How to read and make phylogenetic trees Zuzana Starostová How to make phylogenetic trees? Workflow: obtain DNA sequence quality check sequence alignment calculating genetic distances phylogeny estimation
How to read and make phylogenetic trees Zuzana Starostová How to make phylogenetic trees? Workflow: obtain DNA sequence quality check sequence alignment calculating genetic distances phylogeny estimation
Dr. Amira A. AL-Hosary
 Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Introductory course on Multiple Sequence Alignment Part I: Theoretical foundations
 Sequence Analysis and Structure Prediction Service Centro Nacional de Biotecnología CSIC 8-10 May, 2013 Introductory course on Multiple Sequence Alignment Part I: Theoretical foundations Course Notes Instructor:
Sequence Analysis and Structure Prediction Service Centro Nacional de Biotecnología CSIC 8-10 May, 2013 Introductory course on Multiple Sequence Alignment Part I: Theoretical foundations Course Notes Instructor:
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bio nformatics. Lecture 23. Saad Mneimneh
 Bio nformatics Lecture 23 Protein folding The goal is to determine the three-dimensional structure of a protein based on its amino acid sequence Assumption: amino acid sequence completely and uniquely
Bio nformatics Lecture 23 Protein folding The goal is to determine the three-dimensional structure of a protein based on its amino acid sequence Assumption: amino acid sequence completely and uniquely
p(-,i)+p(,i)+p(-,v)+p(i,v),v)+p(i,v)
+p(,i)+p(-,v)+p(i,v),v)+p(i,v)") Multile Sequence Alignment Given: Set of sequences Score matrix Ga enalties Find: Alignment of sequences such that otimal score is achieved. Motivation Aligning rotein families Establish evolutionary relationshis
Multile Sequence Alignment Given: Set of sequences Score matrix Ga enalties Find: Alignment of sequences such that otimal score is achieved. Motivation Aligning rotein families Establish evolutionary relationshis
Practical considerations of working with sequencing data
 Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Phylogenetics: Bayesian Phylogenetic Analysis. COMP Spring 2015 Luay Nakhleh, Rice University
 Phylogenetics: Bayesian Phylogenetic Analysis COMP 571 - Spring 2015 Luay Nakhleh, Rice University Bayes Rule P(X = x Y = y) = P(X = x, Y = y) P(Y = y) = P(X = x)p(y = y X = x) P x P(X = x 0 )P(Y = y X
Phylogenetics: Bayesian Phylogenetic Analysis COMP 571 - Spring 2015 Luay Nakhleh, Rice University Bayes Rule P(X = x Y = y) = P(X = x, Y = y) P(Y = y) = P(X = x)p(y = y X = x) P x P(X = x 0 )P(Y = y X
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
What is Phylogenetics
 What is Phylogenetics Phylogenetics is the area of research concerned with finding the genetic connections and relationships between species. The basic idea is to compare specific characters (features)
What is Phylogenetics Phylogenetics is the area of research concerned with finding the genetic connections and relationships between species. The basic idea is to compare specific characters (features)
Introduction to Bioinformatics Introduction to Bioinformatics
 Dr. rer. nat. Gong Jing Cancer Research Center Medicine School of Shandong University 2012.11.09 1 Chapter 4 Phylogenetic Tree 2 Phylogeny Evidence from morphological ( 形态学的 ), biochemical, and gene sequence
Dr. rer. nat. Gong Jing Cancer Research Center Medicine School of Shandong University 2012.11.09 1 Chapter 4 Phylogenetic Tree 2 Phylogeny Evidence from morphological ( 形态学的 ), biochemical, and gene sequence
BMI/CS 776 Lecture #20 Alignment of whole genomes. Colin Dewey (with slides adapted from those by Mark Craven)
") BMI/CS 776 Lecture #20 Alignment of whole genomes Colin Dewey (with slides adapted from those by Mark Craven) 2007.03.29 1 Multiple whole genome alignment Input set of whole genome sequences genomes diverged
BMI/CS 776 Lecture #20 Alignment of whole genomes Colin Dewey (with slides adapted from those by Mark Craven) 2007.03.29 1 Multiple whole genome alignment Input set of whole genome sequences genomes diverged
Multiple Alignment. Anders Gorm Pedersen Molecular Evolution Group Center for Biological Sequence Analysis
 Multiple Alignment Anders Gorm Pedersen Molecular Evolution Group Center for Biological Sequence Analysis gorm@cbs.dtu.dk Refresher: pairwise alignments 43.2% identity; Global alignment score: 374 10 20
Multiple Alignment Anders Gorm Pedersen Molecular Evolution Group Center for Biological Sequence Analysis gorm@cbs.dtu.dk Refresher: pairwise alignments 43.2% identity; Global alignment score: 374 10 20
Phylogenetic Trees. What They Are Why We Do It & How To Do It. Presented by Amy Harris Dr Brad Morantz
 Phylogenetic Trees What They Are Why We Do It & How To Do It Presented by Amy Harris Dr Brad Morantz Overview What is a phylogenetic tree Why do we do it How do we do it Methods and programs Parallels
Phylogenetic Trees What They Are Why We Do It & How To Do It Presented by Amy Harris Dr Brad Morantz Overview What is a phylogenetic tree Why do we do it How do we do it Methods and programs Parallels
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Phylogenetics: Building Phylogenetic Trees
 1 Phylogenetics: Building Phylogenetic Trees COMP 571 Luay Nakhleh, Rice University 2 Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary model should
1 Phylogenetics: Building Phylogenetic Trees COMP 571 Luay Nakhleh, Rice University 2 Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary model should
Sequence Alignments. Dynamic programming approaches, scoring, and significance. Lucy Skrabanek ICB, WMC January 31, 2013
 Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
A profile-based protein sequence alignment algorithm for a domain clustering database
 A profile-based protein sequence alignment algorithm for a domain clustering database Lin Xu,2 Fa Zhang and Zhiyong Liu 3, Key Laboratory of Computer System and architecture, the Institute of Computing
A profile-based protein sequence alignment algorithm for a domain clustering database Lin Xu,2 Fa Zhang and Zhiyong Liu 3, Key Laboratory of Computer System and architecture, the Institute of Computing
Mul$ple Sequence Alignment Methods. Tandy Warnow Departments of Bioengineering and Computer Science h?p://tandy.cs.illinois.edu
 Mul$ple Sequence Alignment Methods Tandy Warnow Departments of Bioengineering and Computer Science h?p://tandy.cs.illinois.edu Species Tree Orangutan Gorilla Chimpanzee Human From the Tree of the Life
Mul$ple Sequence Alignment Methods Tandy Warnow Departments of Bioengineering and Computer Science h?p://tandy.cs.illinois.edu Species Tree Orangutan Gorilla Chimpanzee Human From the Tree of the Life
Bioinformatics. Dept. of Computational Biology & Bioinformatics
 Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Phylogeny: traditional and Bayesian approaches
 Phylogeny: traditional and Bayesian approaches 5-Feb-2014 DEKM book Notes from Dr. B. John Holder and Lewis, Nature Reviews Genetics 4, 275-284, 2003 1 Phylogeny A graph depicting the ancestor-descendent
Phylogeny: traditional and Bayesian approaches 5-Feb-2014 DEKM book Notes from Dr. B. John Holder and Lewis, Nature Reviews Genetics 4, 275-284, 2003 1 Phylogeny A graph depicting the ancestor-descendent
Session 5: Phylogenomics
 Session 5: Phylogenomics B.- Phylogeny based orthology assignment REMINDER: Gene tree reconstruction is divided in three steps: homology search, multiple sequence alignment and model selection plus tree
Session 5: Phylogenomics B.- Phylogeny based orthology assignment REMINDER: Gene tree reconstruction is divided in three steps: homology search, multiple sequence alignment and model selection plus tree
Research Proposal. Title: Multiple Sequence Alignment used to investigate the co-evolving positions in OxyR Protein family.
 Research Proposal Title: Multiple Sequence Alignment used to investigate the co-evolving positions in OxyR Protein family. Name: Minjal Pancholi Howard University Washington, DC. June 19, 2009 Research
Research Proposal Title: Multiple Sequence Alignment used to investigate the co-evolving positions in OxyR Protein family. Name: Minjal Pancholi Howard University Washington, DC. June 19, 2009 Research
Overview Multiple Sequence Alignment
 Overview Multiple Sequence Alignment Inge Jonassen Bioinformatics group Dept. of Informatics, UoB Inge.Jonassen@ii.uib.no Definition/examples Use of alignments The alignment problem scoring alignments
Overview Multiple Sequence Alignment Inge Jonassen Bioinformatics group Dept. of Informatics, UoB Inge.Jonassen@ii.uib.no Definition/examples Use of alignments The alignment problem scoring alignments
Phylogenetics: Building Phylogenetic Trees. COMP Fall 2010 Luay Nakhleh, Rice University
 Phylogenetics: Building Phylogenetic Trees COMP 571 - Fall 2010 Luay Nakhleh, Rice University Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary
Phylogenetics: Building Phylogenetic Trees COMP 571 - Fall 2010 Luay Nakhleh, Rice University Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary
Copyright notice. Multiple sequence alignment. Multiple sequence alignment: outline. Multiple sequence alignment: today s goals
 Copyright notice Multiple sequence alignment Monday, December 8, 2008 Many of the images in this powerpoint presentation are from Bioinformatics and Functional Genomics by J Pevsner (ISBN 0-471-21004-8).
Copyright notice Multiple sequence alignment Monday, December 8, 2008 Many of the images in this powerpoint presentation are from Bioinformatics and Functional Genomics by J Pevsner (ISBN 0-471-21004-8).