molecular evolution and phylogenetics
|
|
|
- Rosa Merritt
- 5 years ago
- Views:
Transcription

1 molecular evolution and phylogenetics Charlotte Darby Computational Genomics: Applied Comparative Genomics
2 Internal node Root TIME Branch Leaves A B C D
3 computer scientist biologist??? Each set of three figures shows the same topological relationship among the leaves C E D B A B C D E A A B C D E

4 Internal node Root Leaves Branch Leaves A B C D Strains or isolates of the same species OR different species OR DNA or protein sequences From the same genome or different genomes Leaves are implied to be extant (present at the current time)

5 Internal node Root Root Branch Leaves A B C D Implies what happened first in chronological time Tree-building algorithms may not infer the root of a tree Represented as a node with 3 outgoing branches where the root ought to be Could root halfway along the longest path midpoint rooting Could root by prior knowledge of an outgroup e.g. bacteria in the tree above
6
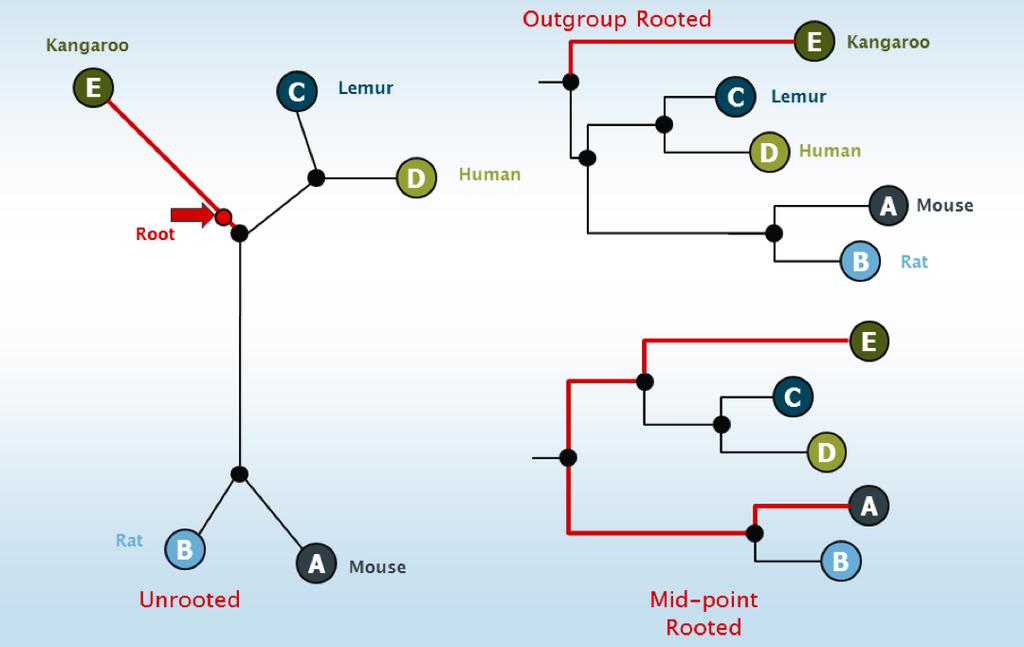
7
 of the leaves following it Internal nodes are implied to NOT be")
8 Internal node Root Internal node Branch Leaves A B C D Represents some ancestral state Most recent common ancestor (MRCA) of the leaves following it Internal nodes are implied to NOT be extant
9 Internal node Root Branch length Branch Leaves A B C D Chronological time OR number of changes to the DNA / protein sequence Depending on assumptions you make about rate of evolution when building tree, may not be ultrametric (every root-leaf path has same length) OR no distance implied just defines a branching order B C A D A B C D
10 Why visualize biological information as trees? (besides having a colorful Figure 1 for your Nature paper!) Trees show fundamental structure in your data. A picture s worth a thousand words!
11 The last amniote common ancestor of synapsids and sauropsids is placed 315 MYA. The sauropsid lineage evolved into archosaurs (such as birds, crocodilians, and dinosaurs) and lepidosaurs (such as snakes and lizards), diverging in the Triassic period. In the synapsid lineage, these primitive mammals acquired homothermy and lactation before the divergence of protetherian and therian mammals 166 MYA in the Jurassic period. While descendant species of the prototherian mammals - extant monotremes such as the platypus Ornithorhynchus anatinus - have characteristics such as venom, electroreception, and meroblastic cleavage, therian mammals evolved holoblastic cleavage, placentation, viviparity, and testicular descent before their divergence 148 MYA into marsupials and eutherians. Further diversification leads us to observe a pouch and prolonged lactation in extant marsupials, and inner cell mass and prolonged gestation in eutherians. Text composed by me for illustration only
 species, and when in evolution did they arise?")
12 species phylogeny When did species diverge? What characteristics define extant (surviving) species, and when in evolution did they arise? Genome analysis of the platypus reveals unique signatures of evolution. Warren et al., Nature 2008

13 strain phylogeny When were pathogens transmitted between individuals, species, or places? Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Faria et al., Nature 2017
14 Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Faria et al., Nature 2017
15 gene phylogeny D D D D D D How did a gene family diversify through duplication? How are the current functions of genes with shared evolutionary history (AKA gene family or homologous genes) related to their evolution? D D Gene duplication event
![(((A,B),C),(D,E)) The Newick Standard was adopted 26 June 1986 by an informal committee meeting convened by me [Joe Felsenstein] during the Society for the Study of Evolution meetings in Durham, New](/docs-images/83/88939556/images/16-0.jpg "Hampshire and consisting of James Archie, William H.E. Day, Wayne Maddison, Christopher Meacham, F. James Rohlf, David Swofford, and myself.")
16 (((A,B),C),(D,E)) The Newick Standard was adopted 26 June 1986 by an informal committee meeting convened by me [Joe Felsenstein] during the Society for the Study of Evolution meetings in Durham, New Hampshire and consisting of James Archie, William H.E. Day, Wayne Maddison, Christopher Meacham, F. James Rohlf, David Swofford, and myself. (The committee was not an activity of the SSE nor endorsed by it). The reason for the name is that the second and final session of the committee met at Newick's restaurant in Dover, New Hampshire, and we enjoyed the meal of lobsters. ((raccoon: ,bear: ): [50],((sea_lion: , seal: ): [100],((monkey: ,cat: ): [80], weasel: ): [75]): [50],dog: ); (((A:5,B:5)f:3,C:9)g:1,(D:2,E:6)h:2);
17 Demo:
18 Building Trees
19 You need a principled way of choosing the tree that best represents the relationship among leaves There are (2n-3)!! rooted trees for n taxa 3 taxa: 3 trees 5 taxa: 105 trees 10 taxa: 34,459,425 trees 20 taxa: trees 50 taxa: trees 100 taxa: trees The double factorial, symbolized by two exclamation marks (!!), is a quantity defined for all integers greater than or equal to -1. For an even integer n, the double factorial is the product of all even integers less than or equal to n but greater than or equal to 2.
20 Parsimony: scenario that requires the fewest changes is the best
21
22 Clustering a distance matrix Calculate all pairwise distances Use a mathematical model of protein / nucleotide evolution to calculate the distance between two sequences Here: Hamming distance Example algorithms: Least squares Neighbor joining Minimum evolution UPGMA (Unweighted Pair Group Method with Arithmetic Mean) Software: PHYLIP AATG AAAG 3A>3T 2A>2C 4G>4T AAAT AAAG ACTG I II III I II 1-2 III 3 2 -

23 Estimating distance between sequences: nucleotide evolution models The thickness of the arrows indicates the substitution rates of the four nucleotides (T, C, A and G), and the sizes of the circles represent the nucleotide frequencies when the substitution process is in equilibrium. Note that both JC69 and K80 predict equal proportions of the four nucleotides. Molecular phylogenetics: principles and practice. Yang and Rannala, Nature Reviews Genetics 2012
 and join those leaves with a new internal node, with branch lengths based on the values in")

24 Clustering: Neighbor joining 0. Start with a star graph 1. Calculate distance matrix 2. Find the minimum entry (closest leaves) and join those leaves with a new internal node, with branch lengths based on the values in the matrix 3. Replace the rows/columns of the leaves with a single row/column of the new internal node 4. Recalculate distance matrix Repeat 2,3,4 until you have an unrooted binary tree *Resulting tree is unrooted and not guaranteed to be ultrametric (Step 2)
 and join those leaves at a distance of the mean distance between their elements 3.")

25 Clustering: UPGMA 0. Start with a star graph 1. Calculate distance matrix 2. Find the minimum entry (closest leaves) and join those leaves at a distance of the mean distance between their elements 3. Replace the rows/columns of the leaves with a single row/column of their union (cluster) 4. Recalculate distance matrix Repeat 2,3,4 until you have an rooted binary tree *Resulting tree is rooted and ultrametric: assumes rate of evolution is constant on all branches (molecular clock)
26

27 Multiple sequence alignment
28 Maximum likelihood estimation from an MSA 0. Start with a multiple sequence alignment 1. Build a starting tree 2. Calculate the likelihood of the tree based on an evolutionary model 3. Change the tree slightly to increase the likelihood Repeat 2,3 until some convergence criterion met ML is a heuristic search of tree space Bootstrapping: resampling from tree space to gauge the quality of your solution Proportion of resamples that support a certain branching pattern Software: PhyML/RAxML Genetic algorithm - GARLI
29 Bayesian estimation from an MSA 0. Start with a multiple sequence alignment 1. Build a starting tree and assume some prior probability on the distribution of trees 2. Calculate the posterior probability of the tree based on an evolutionary model 3. Sample a nearby tree, usually using MCMC (Markov chain Monte Carlo) algorithms 4. Accept the new tree if it is better, otherwise keep with the old tree Repeat 2,3,4 until some convergence criterion met Bayesian methods are also a heuristic search of tree space The posterior probability (probability correct given model) gauges the quality of your solution Software: BEAST, MrBayes

30 Molecular phylogenetics: principles and practice. Yang and Rannala, Nature Reviews Genetics 2012
31 3-hydroxy-3-methylglutaryl coenzyme A synthase (HMGCS) enzyme family Homologs: Genes with shared evolutionary history Orthologs: Genes that diverged through speciation (i.e. not duplication) Paralogs: Genes that diverged through duplication Orthologs Gene duplication event Paralogs Same species Paralogs Different species When type `1` genes cluster separately from type `2` genes in the gene tree, this suggests there was a duplication and that type `1` genes and type `2` genes are paralogs. How old is my gene? Capra et al., Trends in Genetics 2013
32 3-hydroxy-3-methylglutaryl coenzyme A synthase (HMGCS) enzyme family 1 (duplication) amniotes 1,2 fish frog shark 1 More parsimonious Supported by gene tree evidence X X X Gene duplication at root 1,2 (3 losses) Gene tree inferred using PhyML from sequences aligned with MAFFT and rooted using three invertebrate outgroup sequences. Branch support was assessed using alrt scores. How old is my gene? Capra et al., Trends in Genetics 2013 amniotes 1,2 fish frog shark 1
33 Evolution of chromosomes in hexaploid sweet potato a) Evolutionary history of cultivated I. batatas revealed by phylogenetic analysis of homologous chromosome regions. a, The dominant topology structure of all phylogenetic trees. Numbers indicate the average branch length of trees in this structure. b) The time points of B2 subgenome specialization and two whole-genome duplication events were estimated as 1.3, 0.8 and 0.5 million years ago (Ma). The estimation is based on 0.7% mutation rate per million years. The dashed curve indicates the crossing between diploid and tetraploid progenitors. Haplotype-resolved sweet potato genome traces back its hexaploidization history. Yang et. al, Nature Plants 2017

34 Building a species phylogeny Macroscopic characteristics Molecular characteristics Conserved sequence, e.g. ribosome Concatenation of conserved genes Consensus of many individual-gene trees (supertree)
35 Species phylogeny of Cyanobacteria from consensus of many protein families Consensus cyanobacterial phylogeny based on maximum likelihood trees for each of 438 orthologous protein families. The numbers at each bifurcation indicate the total number of trees where that exact bifurcation/branching order is observed Note that as this is a consensus tree, topology is meaningful but distances are not. Integrating Markov Clustering and Molecular Phylogenetics to Reconstruct the Cyanobacterial Species Tree from Conserved Protein Families. Swingley et al., Mol Bio Evol 2008
36 Species phylogeny of Cyanobacteria from concatenation of many multiple sequence alignments [MrBayes] tree for the full-concatenated data set, based on 230,415 aligned positions in 26 genomes. The scale bar indicates the number of substitutions per site. Shown at each bifurcation are the predicted coregenome (upper number) and pan-genome (lower number) sizes of an ancestor at that point. Integrating Markov Clustering and Molecular Phylogenetics to Reconstruct the Cyanobacterial Species Tree from Conserved Protein Families. Swingley et al., Mol Bio Evol 2008
37 + extant species w/ nitrogen fixation X loss of nitrogen fixation if we assume present at root and parallel loss (gray square) ancestor had nitrogen fixation if we assume multiple gains + nitrogen fixation gained on branch if we assume multiple gains More parsimonious: 3 Gains or 5 Losses? Integrating Markov Clustering and Molecular Phylogenetics to Reconstruct the Cyanobacterial Species Tree from Conserved Protein Families. Swingley et al., Mol Bio Evol 2008
38 Resources Review article Molecular phylogenetics: principles and practice. Yang and Rannala, Nature Reviews Genetics 2012 Opinion piece Homology: a personal view on some of the problems. Fitch, Trends In Genetics 2000 Textbooks Molecular Evolution: A Phylogenetic Approach. Page and Holmes Inferring Phylogenies. Felsenstein My work in evolutionary biology (horizontal gene transfer) Xenolog classification. Darby, Stolzer et. al., Bioinformatics 2017
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Distance Methods Character Methods
Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Distance Methods Character Methods
Phylogenetic Tree Reconstruction
 I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
BINF6201/8201. Molecular phylogenetic methods
 BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
Phylogenetic inference
 Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Using phylogenetics to estimate species divergence times... Basics and basic issues for Bayesian inference of divergence times (plus some digression)
") Using phylogenetics to estimate species divergence times... More accurately... Basics and basic issues for Bayesian inference of divergence times (plus some digression) "A comparison of the structures
Using phylogenetics to estimate species divergence times... More accurately... Basics and basic issues for Bayesian inference of divergence times (plus some digression) "A comparison of the structures
EVOLUTIONARY DISTANCES
 EVOLUTIONARY DISTANCES FROM STRINGS TO TREES Luca Bortolussi 1 1 Dipartimento di Matematica ed Informatica Università degli studi di Trieste luca@dmi.units.it Trieste, 14 th November 2007 OUTLINE 1 STRINGS:
EVOLUTIONARY DISTANCES FROM STRINGS TO TREES Luca Bortolussi 1 1 Dipartimento di Matematica ed Informatica Università degli studi di Trieste luca@dmi.units.it Trieste, 14 th November 2007 OUTLINE 1 STRINGS:
Phylogeny: building the tree of life
 Phylogeny: building the tree of life Dr. Fayyaz ul Amir Afsar Minhas Department of Computer and Information Sciences Pakistan Institute of Engineering & Applied Sciences PO Nilore, Islamabad, Pakistan
Phylogeny: building the tree of life Dr. Fayyaz ul Amir Afsar Minhas Department of Computer and Information Sciences Pakistan Institute of Engineering & Applied Sciences PO Nilore, Islamabad, Pakistan
Evolutionary Tree Analysis. Overview
 CSI/BINF 5330 Evolutionary Tree Analysis Young-Rae Cho Associate Professor Department of Computer Science Baylor University Overview Backgrounds Distance-Based Evolutionary Tree Reconstruction Character-Based
CSI/BINF 5330 Evolutionary Tree Analysis Young-Rae Cho Associate Professor Department of Computer Science Baylor University Overview Backgrounds Distance-Based Evolutionary Tree Reconstruction Character-Based
What is Phylogenetics
 What is Phylogenetics Phylogenetics is the area of research concerned with finding the genetic connections and relationships between species. The basic idea is to compare specific characters (features)
What is Phylogenetics Phylogenetics is the area of research concerned with finding the genetic connections and relationships between species. The basic idea is to compare specific characters (features)
9/30/11. Evolution theory. Phylogenetic Tree Reconstruction. Phylogenetic trees (binary trees) Phylogeny (phylogenetic tree)
 Phylogeny (phylogenetic tree)") I9 Introduction to Bioinformatics, 0 Phylogenetic ree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & omputing, IUB Evolution theory Speciation Evolution of new organisms is driven by
I9 Introduction to Bioinformatics, 0 Phylogenetic ree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & omputing, IUB Evolution theory Speciation Evolution of new organisms is driven by
Bioinformatics tools for phylogeny and visualization. Yanbin Yin
 Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
Phylogenetic Trees. Phylogenetic Trees Five. Phylogeny: Inference Tool. Phylogeny Terminology. Picture of Last Quagga. Importance of Phylogeny 5.
 Five Sami Khuri Department of Computer Science San José State University San José, California, USA sami.khuri@sjsu.edu v Distance Methods v Character Methods v Molecular Clock v UPGMA v Maximum Parsimony
Five Sami Khuri Department of Computer Science San José State University San José, California, USA sami.khuri@sjsu.edu v Distance Methods v Character Methods v Molecular Clock v UPGMA v Maximum Parsimony
8/23/2014. Phylogeny and the Tree of Life
 Phylogeny and the Tree of Life Chapter 26 Objectives Explain the following characteristics of the Linnaean system of classification: a. binomial nomenclature b. hierarchical classification List the major
Phylogeny and the Tree of Life Chapter 26 Objectives Explain the following characteristics of the Linnaean system of classification: a. binomial nomenclature b. hierarchical classification List the major
Molecular phylogeny How to infer phylogenetic trees using molecular sequences
 Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 2009 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 2009 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Molecular phylogeny How to infer phylogenetic trees using molecular sequences
 Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 200 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 200 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Constructing Evolutionary/Phylogenetic Trees
 Constructing Evolutionary/Phylogenetic Trees 2 broad categories: istance-based methods Ultrametric Additive: UPGMA Transformed istance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
Constructing Evolutionary/Phylogenetic Trees 2 broad categories: istance-based methods Ultrametric Additive: UPGMA Transformed istance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
Phylogenetic Analysis. Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center
 Phylogenetic Analysis Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center Outline Basic Concepts Tree Construction Methods Distance-based methods
Phylogenetic Analysis Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center Outline Basic Concepts Tree Construction Methods Distance-based methods
 Tree of Life iological Sequence nalysis Chapter http://tolweb.org/tree/ Phylogenetic Prediction ll organisms on Earth have a common ancestor. ll species are related. The relationship is called a phylogeny
Tree of Life iological Sequence nalysis Chapter http://tolweb.org/tree/ Phylogenetic Prediction ll organisms on Earth have a common ancestor. ll species are related. The relationship is called a phylogeny
UoN, CAS, DBSC BIOL102 lecture notes by: Dr. Mustafa A. Mansi. The Phylogenetic Systematics (Phylogeny and Systematics)
") - Phylogeny? - Systematics? The Phylogenetic Systematics (Phylogeny and Systematics) - Phylogenetic systematics? Connection between phylogeny and classification. - Phylogenetic systematics informs the
- Phylogeny? - Systematics? The Phylogenetic Systematics (Phylogeny and Systematics) - Phylogenetic systematics? Connection between phylogeny and classification. - Phylogenetic systematics informs the
Elements of Bioinformatics 14F01 TP5 -Phylogenetic analysis
 Elements of Bioinformatics 14F01 TP5 -Phylogenetic analysis 10 December 2012 - Corrections - Exercise 1 Non-vertebrate chordates generally possess 2 homologs, vertebrates 3 or more gene copies; a Drosophila
Elements of Bioinformatics 14F01 TP5 -Phylogenetic analysis 10 December 2012 - Corrections - Exercise 1 Non-vertebrate chordates generally possess 2 homologs, vertebrates 3 or more gene copies; a Drosophila
"Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky
 MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut
 Faculty of Veterinary Medicine Assiut") Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Chapter 26: Phylogeny and the Tree of Life Phylogenies Show Evolutionary Relationships
 Chapter 26: Phylogeny and the Tree of Life You Must Know The taxonomic categories and how they indicate relatedness. How systematics is used to develop phylogenetic trees. How to construct a phylogenetic
Chapter 26: Phylogeny and the Tree of Life You Must Know The taxonomic categories and how they indicate relatedness. How systematics is used to develop phylogenetic trees. How to construct a phylogenetic
Dr. Amira A. AL-Hosary
 Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Phylogenetic trees 07/10/13
 Phylogenetic trees 07/10/13 A tree is the only figure to occur in On the Origin of Species by Charles Darwin. It is a graphical representation of the evolutionary relationships among entities that share
Phylogenetic trees 07/10/13 A tree is the only figure to occur in On the Origin of Species by Charles Darwin. It is a graphical representation of the evolutionary relationships among entities that share
Phylogenetics: Building Phylogenetic Trees
 1 Phylogenetics: Building Phylogenetic Trees COMP 571 Luay Nakhleh, Rice University 2 Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary model should
1 Phylogenetics: Building Phylogenetic Trees COMP 571 Luay Nakhleh, Rice University 2 Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary model should
Multiple Sequence Alignment. Sequences
 Multiple Sequence Alignment Sequences > YOR020c mstllksaksivplmdrvlvqrikaqaktasglylpe knveklnqaevvavgpgftdangnkvvpqvkvgdqvl ipqfggstiklgnddevilfrdaeilakiakd > crassa mattvrsvksliplldrvlvqrvkaeaktasgiflpe
Multiple Sequence Alignment Sequences > YOR020c mstllksaksivplmdrvlvqrikaqaktasglylpe knveklnqaevvavgpgftdangnkvvpqvkvgdqvl ipqfggstiklgnddevilfrdaeilakiakd > crassa mattvrsvksliplldrvlvqrvkaeaktasgiflpe
C3020 Molecular Evolution. Exercises #3: Phylogenetics
 C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
How to read and make phylogenetic trees Zuzana Starostová
 How to read and make phylogenetic trees Zuzana Starostová How to make phylogenetic trees? Workflow: obtain DNA sequence quality check sequence alignment calculating genetic distances phylogeny estimation
How to read and make phylogenetic trees Zuzana Starostová How to make phylogenetic trees? Workflow: obtain DNA sequence quality check sequence alignment calculating genetic distances phylogeny estimation
Molecular phylogeny - Using molecular sequences to infer evolutionary relationships. Tore Samuelsson Feb 2016
 Molecular phylogeny - Using molecular sequences to infer evolutionary relationships Tore Samuelsson Feb 2016 Molecular phylogeny is being used in the identification and characterization of new pathogens,
Molecular phylogeny - Using molecular sequences to infer evolutionary relationships Tore Samuelsson Feb 2016 Molecular phylogeny is being used in the identification and characterization of new pathogens,
Reading for Lecture 13 Release v10
 Reading for Lecture 13 Release v10 Christopher Lee November 15, 2011 Contents 1 Evolutionary Trees i 1.1 Evolution as a Markov Process...................................... ii 1.2 Rooted vs. Unrooted Trees........................................
Reading for Lecture 13 Release v10 Christopher Lee November 15, 2011 Contents 1 Evolutionary Trees i 1.1 Evolution as a Markov Process...................................... ii 1.2 Rooted vs. Unrooted Trees........................................
Phylogenetics: Building Phylogenetic Trees. COMP Fall 2010 Luay Nakhleh, Rice University
 Phylogenetics: Building Phylogenetic Trees COMP 571 - Fall 2010 Luay Nakhleh, Rice University Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary
Phylogenetics: Building Phylogenetic Trees COMP 571 - Fall 2010 Luay Nakhleh, Rice University Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary
A (short) introduction to phylogenetics
 introduction to phylogenetics") A (short) introduction to phylogenetics Thibaut Jombart, Marie-Pauline Beugin MRC Centre for Outbreak Analysis and Modelling Imperial College London Genetic data analysis with PR Statistics, Millport Field
A (short) introduction to phylogenetics Thibaut Jombart, Marie-Pauline Beugin MRC Centre for Outbreak Analysis and Modelling Imperial College London Genetic data analysis with PR Statistics, Millport Field
Phylogenetics. BIOL 7711 Computational Bioscience
 Consortium for Comparative Genomics! University of Colorado School of Medicine Phylogenetics BIOL 7711 Computational Bioscience Biochemistry and Molecular Genetics Computational Bioscience Program Consortium
Consortium for Comparative Genomics! University of Colorado School of Medicine Phylogenetics BIOL 7711 Computational Bioscience Biochemistry and Molecular Genetics Computational Bioscience Program Consortium
Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction. Lesser Tenrec (Echinops telfairi)
") Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction Lesser Tenrec (Echinops telfairi) Goals: 1. Use phylogenetic experimental design theory to select optimal taxa to
Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction Lesser Tenrec (Echinops telfairi) Goals: 1. Use phylogenetic experimental design theory to select optimal taxa to
Many of the slides that I ll use have been borrowed from Dr. Paul Lewis, Dr. Joe Felsenstein. Thanks!
 Many of the slides that I ll use have been borrowed from Dr. Paul Lewis, Dr. Joe Felsenstein. Thanks! Paul has many great tools for teaching phylogenetics at his web site: http://hydrodictyon.eeb.uconn.edu/people/plewis
Many of the slides that I ll use have been borrowed from Dr. Paul Lewis, Dr. Joe Felsenstein. Thanks! Paul has many great tools for teaching phylogenetics at his web site: http://hydrodictyon.eeb.uconn.edu/people/plewis
Phylogeny and Evolution. Gina Cannarozzi ETH Zurich Institute of Computational Science
 Phylogeny and Evolution Gina Cannarozzi ETH Zurich Institute of Computational Science History Aristotle (384-322 BC) classified animals. He found that dolphins do not belong to the fish but to the mammals.
Phylogeny and Evolution Gina Cannarozzi ETH Zurich Institute of Computational Science History Aristotle (384-322 BC) classified animals. He found that dolphins do not belong to the fish but to the mammals.
METHODS FOR DETERMINING PHYLOGENY. In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task.
 Chapter 12 (Strikberger) Molecular Phylogenies and Evolution METHODS FOR DETERMINING PHYLOGENY In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task. Modern
Chapter 12 (Strikberger) Molecular Phylogenies and Evolution METHODS FOR DETERMINING PHYLOGENY In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task. Modern
Phylogenetic Trees. What They Are Why We Do It & How To Do It. Presented by Amy Harris Dr Brad Morantz
 Phylogenetic Trees What They Are Why We Do It & How To Do It Presented by Amy Harris Dr Brad Morantz Overview What is a phylogenetic tree Why do we do it How do we do it Methods and programs Parallels
Phylogenetic Trees What They Are Why We Do It & How To Do It Presented by Amy Harris Dr Brad Morantz Overview What is a phylogenetic tree Why do we do it How do we do it Methods and programs Parallels
Theory of Evolution Charles Darwin
 Theory of Evolution Charles arwin 858-59: Origin of Species 5 year voyage of H.M.S. eagle (83-36) Populations have variations. Natural Selection & Survival of the fittest: nature selects best adapted varieties
Theory of Evolution Charles arwin 858-59: Origin of Species 5 year voyage of H.M.S. eagle (83-36) Populations have variations. Natural Selection & Survival of the fittest: nature selects best adapted varieties
Phylogeny and the Tree of Life
 Chapter 26 Phylogeny and the Tree of Life PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from
Chapter 26 Phylogeny and the Tree of Life PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from
CHAPTERS 24-25: Evidence for Evolution and Phylogeny
 CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
Chapter 27: Evolutionary Genetics
 Chapter 27: Evolutionary Genetics Student Learning Objectives Upon completion of this chapter you should be able to: 1. Understand what the term species means to biology. 2. Recognize the various patterns
Chapter 27: Evolutionary Genetics Student Learning Objectives Upon completion of this chapter you should be able to: 1. Understand what the term species means to biology. 2. Recognize the various patterns
Phylogeny and the Tree of Life
 Chapter 26 Phylogeny and the Tree of Life PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from
Chapter 26 Phylogeny and the Tree of Life PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from
C.DARWIN ( )
") C.DARWIN (1809-1882) LAMARCK Each evolutionary lineage has evolved, transforming itself, from a ancestor appeared by spontaneous generation DARWIN All organisms are historically interconnected. Their relationships
C.DARWIN (1809-1882) LAMARCK Each evolutionary lineage has evolved, transforming itself, from a ancestor appeared by spontaneous generation DARWIN All organisms are historically interconnected. Their relationships
Chapter 26: Phylogeny and the Tree of Life
 Chapter 26: Phylogeny and the Tree of Life 1. Key Concepts Pertaining to Phylogeny 2. Determining Phylogenies 3. Evolutionary History Revealed in Genomes 1. Key Concepts Pertaining to Phylogeny PHYLOGENY
Chapter 26: Phylogeny and the Tree of Life 1. Key Concepts Pertaining to Phylogeny 2. Determining Phylogenies 3. Evolutionary History Revealed in Genomes 1. Key Concepts Pertaining to Phylogeny PHYLOGENY
CS5263 Bioinformatics. Guest Lecture Part II Phylogenetics
 CS5263 Bioinformatics Guest Lecture Part II Phylogenetics Up to now we have focused on finding similarities, now we start focusing on differences (dissimilarities leading to distance measures). Identifying
CS5263 Bioinformatics Guest Lecture Part II Phylogenetics Up to now we have focused on finding similarities, now we start focusing on differences (dissimilarities leading to distance measures). Identifying
Lecture 6 Phylogenetic Inference
 Lecture 6 Phylogenetic Inference From Darwin s notebook in 1837 Charles Darwin Willi Hennig From The Origin in 1859 Cladistics Phylogenetic inference Willi Hennig, Cladistics 1. Clade, Monophyletic group,
Lecture 6 Phylogenetic Inference From Darwin s notebook in 1837 Charles Darwin Willi Hennig From The Origin in 1859 Cladistics Phylogenetic inference Willi Hennig, Cladistics 1. Clade, Monophyletic group,
Phylogeny and systematics. Why are these disciplines important in evolutionary biology and how are they related to each other?
 Phylogeny and systematics Why are these disciplines important in evolutionary biology and how are they related to each other? Phylogeny and systematics Phylogeny: the evolutionary history of a species
Phylogeny and systematics Why are these disciplines important in evolutionary biology and how are they related to each other? Phylogeny and systematics Phylogeny: the evolutionary history of a species
Phylogenetics. Applications of phylogenetics. Unrooted networks vs. rooted trees. Outline
 Phylogenetics Todd Vision iology 522 March 26, 2007 pplications of phylogenetics Studying organismal or biogeographic history Systematics ating events in the fossil record onservation biology Studying
Phylogenetics Todd Vision iology 522 March 26, 2007 pplications of phylogenetics Studying organismal or biogeographic history Systematics ating events in the fossil record onservation biology Studying
POPULATION GENETICS Winter 2005 Lecture 17 Molecular phylogenetics
 POPULATION GENETICS Winter 2005 Lecture 17 Molecular phylogenetics - in deriving a phylogeny our goal is simply to reconstruct the historical relationships between a group of taxa. - before we review the
POPULATION GENETICS Winter 2005 Lecture 17 Molecular phylogenetics - in deriving a phylogeny our goal is simply to reconstruct the historical relationships between a group of taxa. - before we review the
Phylogenetic Analysis
 Phylogenetic Analysis Aristotle Through classification, one might discover the essence and purpose of species. Nelson & Platnick (1981) Systematics and Biogeography Carl Linnaeus Swedish botanist (1700s)
Phylogenetic Analysis Aristotle Through classification, one might discover the essence and purpose of species. Nelson & Platnick (1981) Systematics and Biogeography Carl Linnaeus Swedish botanist (1700s)
Phylogenetic Analysis
 Phylogenetic Analysis Aristotle Through classification, one might discover the essence and purpose of species. Nelson & Platnick (1981) Systematics and Biogeography Carl Linnaeus Swedish botanist (1700s)
Phylogenetic Analysis Aristotle Through classification, one might discover the essence and purpose of species. Nelson & Platnick (1981) Systematics and Biogeography Carl Linnaeus Swedish botanist (1700s)
Phylogenetic Analysis
 Phylogenetic Analysis Aristotle Through classification, one might discover the essence and purpose of species. Nelson & Platnick (1981) Systematics and Biogeography Carl Linnaeus Swedish botanist (1700s)
Phylogenetic Analysis Aristotle Through classification, one might discover the essence and purpose of species. Nelson & Platnick (1981) Systematics and Biogeography Carl Linnaeus Swedish botanist (1700s)
1 ATGGGTCTC 2 ATGAGTCTC
 We need an optimality criterion to choose a best estimate (tree) Other optimality criteria used to choose a best estimate (tree) Parsimony: begins with the assumption that the simplest hypothesis that
We need an optimality criterion to choose a best estimate (tree) Other optimality criteria used to choose a best estimate (tree) Parsimony: begins with the assumption that the simplest hypothesis that
Molecular Evolution & Phylogenetics
 Molecular Evolution & Phylogenetics Heuristics based on tree alterations, maximum likelihood, Bayesian methods, statistical confidence measures Jean-Baka Domelevo Entfellner Learning Objectives know basic
Molecular Evolution & Phylogenetics Heuristics based on tree alterations, maximum likelihood, Bayesian methods, statistical confidence measures Jean-Baka Domelevo Entfellner Learning Objectives know basic
Anatomy of a tree. clade is group of organisms with a shared ancestor. a monophyletic group shares a single common ancestor = tapirs-rhinos-horses
 Anatomy of a tree outgroup: an early branching relative of the interest groups sister taxa: taxa derived from the same recent ancestor polytomy: >2 taxa emerge from a node Anatomy of a tree clade is group
Anatomy of a tree outgroup: an early branching relative of the interest groups sister taxa: taxa derived from the same recent ancestor polytomy: >2 taxa emerge from a node Anatomy of a tree clade is group
Bayesian Models for Phylogenetic Trees
 Bayesian Models for Phylogenetic Trees Clarence Leung* 1 1 McGill Centre for Bioinformatics, McGill University, Montreal, Quebec, Canada ABSTRACT Introduction: Inferring genetic ancestry of different species
Bayesian Models for Phylogenetic Trees Clarence Leung* 1 1 McGill Centre for Bioinformatics, McGill University, Montreal, Quebec, Canada ABSTRACT Introduction: Inferring genetic ancestry of different species
Comparative Genomics II
 Comparative Genomics II Advances in Bioinformatics and Genomics GEN 240B Jason Stajich May 19 Comparative Genomics II Slide 1/31 Outline Introduction Gene Families Pairwise Methods Phylogenetic Methods
Comparative Genomics II Advances in Bioinformatics and Genomics GEN 240B Jason Stajich May 19 Comparative Genomics II Slide 1/31 Outline Introduction Gene Families Pairwise Methods Phylogenetic Methods
Constructing Evolutionary/Phylogenetic Trees
 Constructing Evolutionary/Phylogenetic Trees 2 broad categories: Distance-based methods Ultrametric Additive: UPGMA Transformed Distance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
Constructing Evolutionary/Phylogenetic Trees 2 broad categories: Distance-based methods Ultrametric Additive: UPGMA Transformed Distance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
Comparative Bioinformatics Midterm II Fall 2004
 Comparative Bioinformatics Midterm II Fall 2004 Objective Answer, part I: For each of the following, select the single best answer or completion of the phrase. (3 points each) 1. Deinococcus radiodurans
Comparative Bioinformatics Midterm II Fall 2004 Objective Answer, part I: For each of the following, select the single best answer or completion of the phrase. (3 points each) 1. Deinococcus radiodurans
Phylogenetic analysis. Characters
 Typical steps: Phylogenetic analysis Selection of taxa. Selection of characters. Construction of data matrix: character coding. Estimating the best-fitting tree (model) from the data matrix: phylogenetic
Typical steps: Phylogenetic analysis Selection of taxa. Selection of characters. Construction of data matrix: character coding. Estimating the best-fitting tree (model) from the data matrix: phylogenetic
Integrative Biology 200 "PRINCIPLES OF PHYLOGENETICS" Spring 2018 University of California, Berkeley
 Integrative Biology 200 "PRINCIPLES OF PHYLOGENETICS" Spring 2018 University of California, Berkeley B.D. Mishler Feb. 14, 2018. Phylogenetic trees VI: Dating in the 21st century: clocks, & calibrations;
Integrative Biology 200 "PRINCIPLES OF PHYLOGENETICS" Spring 2018 University of California, Berkeley B.D. Mishler Feb. 14, 2018. Phylogenetic trees VI: Dating in the 21st century: clocks, & calibrations;
Phylogeny. November 7, 2017
 Phylogeny November 7, 2017 Phylogenetics Phylon = tribe/race, genetikos = relative to birth Phylogenetics: study of evolutionary relationships among organisms, sequences, or anything in between Related
Phylogeny November 7, 2017 Phylogenetics Phylon = tribe/race, genetikos = relative to birth Phylogenetics: study of evolutionary relationships among organisms, sequences, or anything in between Related
Phylogenetic analyses. Kirsi Kostamo
 Phylogenetic analyses Kirsi Kostamo The aim: To construct a visual representation (a tree) to describe the assumed evolution occurring between and among different groups (individuals, populations, species,
Phylogenetic analyses Kirsi Kostamo The aim: To construct a visual representation (a tree) to describe the assumed evolution occurring between and among different groups (individuals, populations, species,
Bayesian Inference using Markov Chain Monte Carlo in Phylogenetic Studies
 Bayesian Inference using Markov Chain Monte Carlo in Phylogenetic Studies 1 What is phylogeny? Essay written for the course in Markov Chains 2004 Torbjörn Karfunkel Phylogeny is the evolutionary development
Bayesian Inference using Markov Chain Monte Carlo in Phylogenetic Studies 1 What is phylogeny? Essay written for the course in Markov Chains 2004 Torbjörn Karfunkel Phylogeny is the evolutionary development
Biology 559R: Introduction to Phylogenetic Comparative Methods Topics for this week (Jan 27 & 29):
:") Biology 559R: Introduction to Phylogenetic Comparative Methods Topics for this week (Jan 27 & 29): Statistical estimation of models of sequence evolution Phylogenetic inference using maximum likelihood:
Biology 559R: Introduction to Phylogenetic Comparative Methods Topics for this week (Jan 27 & 29): Statistical estimation of models of sequence evolution Phylogenetic inference using maximum likelihood:
Phylogenetic relationship among S. castellii, S. cerevisiae and C. glabrata.
 Supplementary Note S2 Phylogenetic relationship among S. castellii, S. cerevisiae and C. glabrata. Phylogenetic trees reconstructed by a variety of methods from either single-copy orthologous loci (Class
Supplementary Note S2 Phylogenetic relationship among S. castellii, S. cerevisiae and C. glabrata. Phylogenetic trees reconstructed by a variety of methods from either single-copy orthologous loci (Class
Michael Yaffe Lecture #5 (((A,B)C)D) Database Searching & Molecular Phylogenetics A B C D B C D
C)D) Database Searching & Molecular Phylogenetics A B C D B C D") 7.91 Lecture #5 Database Searching & Molecular Phylogenetics Michael Yaffe B C D B C D (((,B)C)D) Outline Distance Matrix Methods Neighbor-Joining Method and Related Neighbor Methods Maximum Likelihood
7.91 Lecture #5 Database Searching & Molecular Phylogenetics Michael Yaffe B C D B C D (((,B)C)D) Outline Distance Matrix Methods Neighbor-Joining Method and Related Neighbor Methods Maximum Likelihood
Module 12: Molecular Phylogenetics
 Module 12: Molecular Phylogenetics http://evolution.gs.washington.edu/sisg/2014/ MTH Thanks to Paul Lewis, Tracy Heath, Joe Felsenstein, Peter Beerli, Derrick Zwickl, and Joe Bielawski for slides Monday
Module 12: Molecular Phylogenetics http://evolution.gs.washington.edu/sisg/2014/ MTH Thanks to Paul Lewis, Tracy Heath, Joe Felsenstein, Peter Beerli, Derrick Zwickl, and Joe Bielawski for slides Monday
Chapter 26 Phylogeny and the Tree of Life
 Chapter 26 Phylogeny and the Tree of Life Chapter focus Shifting from the process of how evolution works to the pattern evolution produces over time. Phylogeny Phylon = tribe, geny = genesis or origin
Chapter 26 Phylogeny and the Tree of Life Chapter focus Shifting from the process of how evolution works to the pattern evolution produces over time. Phylogeny Phylon = tribe, geny = genesis or origin
Phylogeny: traditional and Bayesian approaches
 Phylogeny: traditional and Bayesian approaches 5-Feb-2014 DEKM book Notes from Dr. B. John Holder and Lewis, Nature Reviews Genetics 4, 275-284, 2003 1 Phylogeny A graph depicting the ancestor-descendent
Phylogeny: traditional and Bayesian approaches 5-Feb-2014 DEKM book Notes from Dr. B. John Holder and Lewis, Nature Reviews Genetics 4, 275-284, 2003 1 Phylogeny A graph depicting the ancestor-descendent
Session 5: Phylogenomics
 Session 5: Phylogenomics B.- Phylogeny based orthology assignment REMINDER: Gene tree reconstruction is divided in three steps: homology search, multiple sequence alignment and model selection plus tree
Session 5: Phylogenomics B.- Phylogeny based orthology assignment REMINDER: Gene tree reconstruction is divided in three steps: homology search, multiple sequence alignment and model selection plus tree
Chapter 16: Reconstructing and Using Phylogenies
 Chapter Review 1. Use the phylogenetic tree shown at the right to complete the following. a. Explain how many clades are indicated: Three: (1) chimpanzee/human, (2) chimpanzee/ human/gorilla, and (3)chimpanzee/human/
Chapter Review 1. Use the phylogenetic tree shown at the right to complete the following. a. Explain how many clades are indicated: Three: (1) chimpanzee/human, (2) chimpanzee/ human/gorilla, and (3)chimpanzee/human/
Organizing Life s Diversity
 17 Organizing Life s Diversity section 2 Modern Classification Classification systems have changed over time as information has increased. What You ll Learn species concepts methods to reveal phylogeny
17 Organizing Life s Diversity section 2 Modern Classification Classification systems have changed over time as information has increased. What You ll Learn species concepts methods to reveal phylogeny
Seuqence Analysis '17--lecture 10. Trees types of trees Newick notation UPGMA Fitch Margoliash Distance vs Parsimony
 Seuqence nalysis '17--lecture 10 Trees types of trees Newick notation UPGM Fitch Margoliash istance vs Parsimony Phyogenetic trees What is a phylogenetic tree? model of evolutionary relationships -- common
Seuqence nalysis '17--lecture 10 Trees types of trees Newick notation UPGM Fitch Margoliash istance vs Parsimony Phyogenetic trees What is a phylogenetic tree? model of evolutionary relationships -- common
Page 1. Evolutionary Trees. Why build evolutionary tree? Outline
 Page Evolutionary Trees Russ. ltman MI S 7 Outline. Why build evolutionary trees?. istance-based vs. character-based methods. istance-based: Ultrametric Trees dditive Trees. haracter-based: Perfect phylogeny
Page Evolutionary Trees Russ. ltman MI S 7 Outline. Why build evolutionary trees?. istance-based vs. character-based methods. istance-based: Ultrametric Trees dditive Trees. haracter-based: Perfect phylogeny
Gene Families part 2. Review: Gene Families /727 Lecture 8. Protein family. (Multi)gene family
gene family") Review: Gene Families Gene Families part 2 03 327/727 Lecture 8 What is a Case study: ian globin genes Gene trees and how they differ from species trees Homology, orthology, and paralogy Last tuesday 1
Review: Gene Families Gene Families part 2 03 327/727 Lecture 8 What is a Case study: ian globin genes Gene trees and how they differ from species trees Homology, orthology, and paralogy Last tuesday 1
Phylogeny Tree Algorithms
 Phylogeny Tree lgorithms Jianlin heng, PhD School of Electrical Engineering and omputer Science University of entral Florida 2006 Free for academic use. opyright @ Jianlin heng & original sources for some
Phylogeny Tree lgorithms Jianlin heng, PhD School of Electrical Engineering and omputer Science University of entral Florida 2006 Free for academic use. opyright @ Jianlin heng & original sources for some
Module 13: Molecular Phylogenetics
 Module 13: Molecular Phylogenetics Instructors: Joe Felsenstein (University of Washington) Mark Holder (University of Kansas) Jeff Thorne (North Carolina State University) Wednesday July 18: Day I 1:30
Module 13: Molecular Phylogenetics Instructors: Joe Felsenstein (University of Washington) Mark Holder (University of Kansas) Jeff Thorne (North Carolina State University) Wednesday July 18: Day I 1:30
Molecular Phylogenetics (part 1 of 2) Computational Biology Course João André Carriço
 Computational Biology Course João André Carriço") Molecular Phylogenetics (part 1 of 2) Computational Biology Course João André Carriço jcarrico@fm.ul.pt Charles Darwin (1809-1882) Charles Darwin s tree of life in Notebook B, 1837-1838 Ernst Haeckel (1934-1919)
Molecular Phylogenetics (part 1 of 2) Computational Biology Course João André Carriço jcarrico@fm.ul.pt Charles Darwin (1809-1882) Charles Darwin s tree of life in Notebook B, 1837-1838 Ernst Haeckel (1934-1919)
Consensus Methods. * You are only responsible for the first two
 Consensus Trees * consensus trees reconcile clades from different trees * consensus is a conservative estimate of phylogeny that emphasizes points of agreement * philosophy: agreement among data sets is
Consensus Trees * consensus trees reconcile clades from different trees * consensus is a conservative estimate of phylogeny that emphasizes points of agreement * philosophy: agreement among data sets is
Effects of Gap Open and Gap Extension Penalties
 Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Phylogeny and Systematics
 Chapter 25 Phylogeny and Systematics PowerPoint Lectures for Biology, Seventh Edition Neil Campbell and Jane Reece Lectures by Chris Romero Modified by Maria Morlin racing phylogeny Phylogeny: he evolutionary
Chapter 25 Phylogeny and Systematics PowerPoint Lectures for Biology, Seventh Edition Neil Campbell and Jane Reece Lectures by Chris Romero Modified by Maria Morlin racing phylogeny Phylogeny: he evolutionary
A Phylogenetic Network Construction due to Constrained Recombination
 A Phylogenetic Network Construction due to Constrained Recombination Mohd. Abdul Hai Zahid Research Scholar Research Supervisors: Dr. R.C. Joshi Dr. Ankush Mittal Department of Electronics and Computer
A Phylogenetic Network Construction due to Constrained Recombination Mohd. Abdul Hai Zahid Research Scholar Research Supervisors: Dr. R.C. Joshi Dr. Ankush Mittal Department of Electronics and Computer
Cladistics and Bioinformatics Questions 2013
 AP Biology Name Cladistics and Bioinformatics Questions 2013 1. The following table shows the percentage similarity in sequences of nucleotides from a homologous gene derived from five different species
AP Biology Name Cladistics and Bioinformatics Questions 2013 1. The following table shows the percentage similarity in sequences of nucleotides from a homologous gene derived from five different species
Bioinformatics 1. Sepp Hochreiter. Biology, Sequences, Phylogenetics Part 4. Bioinformatics 1: Biology, Sequences, Phylogenetics
 Bioinformatics 1 Biology, Sequences, Phylogenetics Part 4 Sepp Hochreiter Klausur Mo. 30.01.2011 Zeit: 15:30 17:00 Raum: HS14 Anmeldung Kusss Contents Methods and Bootstrapping of Maximum Methods Methods
Bioinformatics 1 Biology, Sequences, Phylogenetics Part 4 Sepp Hochreiter Klausur Mo. 30.01.2011 Zeit: 15:30 17:00 Raum: HS14 Anmeldung Kusss Contents Methods and Bootstrapping of Maximum Methods Methods
MOLECULAR SYSTEMATICS: A SYNTHESIS OF THE COMMON METHODS AND THE STATE OF KNOWLEDGE
 CELLULAR & MOLECULAR BIOLOGY LETTERS http://www.cmbl.org.pl Received: 16 August 2009 Volume 15 (2010) pp 311-341 Final form accepted: 01 March 2010 DOI: 10.2478/s11658-010-0010-8 Published online: 19 March
CELLULAR & MOLECULAR BIOLOGY LETTERS http://www.cmbl.org.pl Received: 16 August 2009 Volume 15 (2010) pp 311-341 Final form accepted: 01 March 2010 DOI: 10.2478/s11658-010-0010-8 Published online: 19 March
Inferring phylogeny. Constructing phylogenetic trees. Tõnu Margus. Bioinformatics MTAT
 Inferring phylogeny Constructing phylogenetic trees Tõnu Margus Contents What is phylogeny? How/why it is possible to infer it? Representing evolutionary relationships on trees What type questions questions
Inferring phylogeny Constructing phylogenetic trees Tõnu Margus Contents What is phylogeny? How/why it is possible to infer it? Representing evolutionary relationships on trees What type questions questions
Chapter 19: Taxonomy, Systematics, and Phylogeny
 Chapter 19: Taxonomy, Systematics, and Phylogeny AP Curriculum Alignment Chapter 19 expands on the topics of phylogenies and cladograms, which are important to Big Idea 1. In order for students to understand
Chapter 19: Taxonomy, Systematics, and Phylogeny AP Curriculum Alignment Chapter 19 expands on the topics of phylogenies and cladograms, which are important to Big Idea 1. In order for students to understand
Phylogeny and the Tree of Life
 LECTURE PRESENTATIONS For CAMPBELL BIOLOGY, NINTH EDITION Jane B. Reece, Lisa A. Urry, Michael L. Cain, Steven A. Wasserman, Peter V. Minorsky, Robert B. Jackson Chapter 26 Phylogeny and the Tree of Life
LECTURE PRESENTATIONS For CAMPBELL BIOLOGY, NINTH EDITION Jane B. Reece, Lisa A. Urry, Michael L. Cain, Steven A. Wasserman, Peter V. Minorsky, Robert B. Jackson Chapter 26 Phylogeny and the Tree of Life
Plan: Evolutionary trees, characters. Perfect phylogeny Methods: NJ, parsimony, max likelihood, Quartet method
 Phylogeny 1 Plan: Phylogeny is an important subject. We have 2.5 hours. So I will teach all the concepts via one example of a chain letter evolution. The concepts we will discuss include: Evolutionary
Phylogeny 1 Plan: Phylogeny is an important subject. We have 2.5 hours. So I will teach all the concepts via one example of a chain letter evolution. The concepts we will discuss include: Evolutionary
Phylogenies Scores for Exhaustive Maximum Likelihood and Parsimony Scores Searches
 Int. J. Bioinformatics Research and Applications, Vol. x, No. x, xxxx Phylogenies Scores for Exhaustive Maximum Likelihood and s Searches Hyrum D. Carroll, Perry G. Ridge, Mark J. Clement, Quinn O. Snell
Int. J. Bioinformatics Research and Applications, Vol. x, No. x, xxxx Phylogenies Scores for Exhaustive Maximum Likelihood and s Searches Hyrum D. Carroll, Perry G. Ridge, Mark J. Clement, Quinn O. Snell
Lecture 11 Friday, October 21, 2011
 Lecture 11 Friday, October 21, 2011 Phylogenetic tree (phylogeny) Darwin and classification: In the Origin, Darwin said that descent from a common ancestral species could explain why the Linnaean system
Lecture 11 Friday, October 21, 2011 Phylogenetic tree (phylogeny) Darwin and classification: In the Origin, Darwin said that descent from a common ancestral species could explain why the Linnaean system
Phylogeny and the Tree of Life
 Chapter 26 Phylogeny and the Tree of Life PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from
Chapter 26 Phylogeny and the Tree of Life PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from
Estimating Evolutionary Trees. Phylogenetic Methods
 Estimating Evolutionary Trees v if the data are consistent with infinite sites then all methods should yield the same tree v it gets more complicated when there is homoplasy, i.e., parallel or convergent
Estimating Evolutionary Trees v if the data are consistent with infinite sites then all methods should yield the same tree v it gets more complicated when there is homoplasy, i.e., parallel or convergent
Molecular Evolution and Phylogenetic Tree Reconstruction
 1 4 Molecular Evolution and Phylogenetic Tree Reconstruction 3 2 5 1 4 2 3 5 Orthology, Paralogy, Inparalogs, Outparalogs Phylogenetic Trees Nodes: species Edges: time of independent evolution Edge length
1 4 Molecular Evolution and Phylogenetic Tree Reconstruction 3 2 5 1 4 2 3 5 Orthology, Paralogy, Inparalogs, Outparalogs Phylogenetic Trees Nodes: species Edges: time of independent evolution Edge length
Chapter 26 Phylogeny and the Tree of Life
 Chapter 26 Phylogeny and the Tree of Life Biologists estimate that there are about 5 to 100 million species of organisms living on Earth today. Evidence from morphological, biochemical, and gene sequence
Chapter 26 Phylogeny and the Tree of Life Biologists estimate that there are about 5 to 100 million species of organisms living on Earth today. Evidence from morphological, biochemical, and gene sequence
7. Tests for selection
 Sequence analysis and genomics 7. Tests for selection Dr. Katja Nowick Group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute for Brain Research www. nowicklab.info
Sequence analysis and genomics 7. Tests for selection Dr. Katja Nowick Group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute for Brain Research www. nowicklab.info