Multiple sequence alignment
|
|
|
- Anis Parker
- 6 years ago
- Views:
Transcription
1 Multiple sequence alignment Wednesday, October 11, 2006 Sarah Wheelan
2 Copyright notice Many of the images in this powerpoint presentation are from Bioinformatics and Functional Genomics by J Pevsner (ISBN ). Copyright 2003 by Wiley. These images and materials may not be used without permission from the publisher. Visit
3 Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple sequence alignments to introduce ways you can make your own multiple sequence alignments to show how a multiple sequence alignment provides the basis for phylogenetic trees Page 319

4 Multiple sequence alignment: definition Wallace, Blackshields, Higgins Curr Opp Struct Biol :
5 Multiple sequence alignment: properties Generally align proteins: nucleotides less well-conserved nucleotide sequences are less informative -> harder to align with high confidence How do you know if you have the correct alignment of a protein family? Is there one correct alignment? for two proteins sharing 30% amino acid identity, about 50% of the individual amino acids are superposable in the two structures Page 320
6 Proportion of structurally superposable residues in pairwise alignments as a function of sequence identity Proportion of residues in common core Globin Cytochrome c Serine protease Immunoglobulin domain Sequence identity (%) After Chothia & Lesk (1986)
7 Multiple sequence alignment: features some aligned residues, such as cysteines that form disulfide bridges, may be highly conserved there may be conserved motifs such as a transmembrane domain there may be conserved secondary structure features there may be regions with consistent patterns of insertions or deletions (indels), often indicating the presence of subfamilies Page 320
8 Multiple sequence alignment: uses MSA is more sensitive than pairwise alignment in detecting homologs Detect conserved motifs and domains (from database search or from alignment of a family) Phylogenetic analysis: MSA provides accurate estimation of evolutionary distances 2 and 3 structure prediction: MSA predictions significantly better than from a single sequence very few bioinformatics protocols bypass MSA stage Page 321
9 Multiple sequence alignment: methods There are four main ways to make a multiple sequence alignment: (1) Exact methods (2) Progressive alignment (CLUSTALW, MUSCLE) (3) Iterative approaches (Praline, IterAlign) (4) Consistency-based methods (ProbCons)

10 Multiple sequence alignment: methods Exact methods: dynamic programming Instead of the 2-D dp matrix that you saw in the Needleman-Wunsch technique, think about a 3-D, 4-D etc matrix. Exact methods give optimal alignments but are not feasible in time or space for more than ~10 sequences Still an extremely active field though the holy grail
11 Multiple sequence alignment: methods Progressive methods: use a guide tree (a little like a phylogenetic tree but NOT a phylogenetic tree) to determine how to combine pairwise alignments one by one to create a multiple alignment. Making multiple alignments using trees was a very popular subject in the 80s. Fitch and Yasunobu (1974) may have first proposed the idea, but Hogeweg and Hesper (1984) and many others worked on the topic before Feng and Doolittle (1987) hit the scene they made one important contribution that got their names attached to this alignment method. Examples: CLUSTALW, MUSCLE
12 Multiple sequence alignment: methods Iterative methods: compute a sub-optimal solution and keep modifying that intelligently using dynamic programming or other methods until the solution converges. Examples: IterAlign, Praline, MAFFT
13 Multiple sequence alignment: methods Consistency-based algorithms: generally use a database of both local high-scoring alignments and long-range global alignments to create a final alignment These are very powerful, very fast, and very accurate methods Examples: T-COFFEE, Prrp, DiAlign, ProbCons
14 Multiple sequence alignment: methods Aaack! This is confusing! How do we know which program to use? There are benchmarking multiple alignment datasets that have been aligned painstakingly by hand, by structural similarity, or by extremely time- and memory-intensive automated exact algorithms. Also, to be practical, some programs have interfaces that are much more user-friendly than others. And most programs are excellent so it depends on your preference. If your proteins have 3D structures, USE THESE to help you judge your alignments.
15 Multiple sequence alignment: methods Benchmarking tests suggest that ProbCons, a consistency-based/progressive algorithm, performs the best on the BAliBASE set, although MUSCLE, a progressive alignment package, is an extremely fast and accurate program. CLUSTALW, everyone s old favorite, continues to be a standout and is included in almost every MSA paper you will see. It has withstood the test of time. Plus, it has a nice interface (expecially with CLUSTALX) and is easy to use.
16 Multiple sequence alignment: methods Example of progressive MSA using CLUSTALW: two data sets Five distantly related lipocalins (human to E. coli) Five closely related RBPs When you do this, obtain the sequences of interest in FASTA format! Page 321

17 Get sequences from GenBank

18 Get sequences from GenBank

19 Use Clustal W to do a progressive MSA
20 Feng-Doolittle MSA occurs in 3 stages [1] Do a set of global pairwise alignments (Needleman and Wunsch) [2] Create a guide tree [3] Progressively align the sequences according to the guide tree Page 321
21 Progressive MSA stage 1 of 3: generate global pairwise alignments five distantly related lipocalins best score
22 Progressive MSA stage 1 of 3: generate global pairwise alignments five closely related lipocalins best score
23 Number of pairwise alignments needed Need to align every sequence to every other sequence (handshake problem) For N sequences, (N-1)(N)/2 For 5 sequences, (4)(5)/2 = 10 Page 322
24 Feng-Doolittle stage 2: guide tree Convert similarity scores to distance scores (see text) A guide tree shows the distance between objects Use UPGMA (defined in Chapter 11) ClustalW provides a syntax to describe the tree A guide tree is not a phylogenetic tree Page 323
25 Progressive MSA stage 2 of 3: generate a guide tree calculated from the distance matrix five distantly related lipocalins
26 Progressive MSA stage 2 of 3: generate guide tree five closely related lipocalins
27 Feng-Doolittle stage 3: progressive alignment Make a MSA based on the order in the guide tree Start with the two most closely related sequences Then add the next closest sequence Continue until all sequences are added to the MSA Rule: once a gap, always a gap. <- this is what made them famous! Page 324
28 Why once a gap, always a gap? There are many possible ways to make a MSA Where gaps are added is a critical question Gaps are first added to the first two (closest) sequences To change the initial gap choices later on would be to give more weight to distantly related sequences To maintain the initial gap choices is to trust that those gaps are most believable (shouldn t let the final alignment be affected most by distantly related sequences!) Page 324


29 Progressive MSA stage 3 of 3: progressively align the sequences following the branch order of the tree Fig Page 324

30 Progressive MSA stage 3 of 3: CLUSTALX output
31 Clustal W alignment of 5 closely related lipocalins CLUSTAL W (1.82) multiple sequence alignment gi pir A39486 MEWVWALVLLAALGSAQAERDCRVSSFRVKENFDKARFSGTWYAMAKKDP 50 gi sp P18902 RETB_BOVIN ERDCRVSSFRVKENFDKARFAGTWYAMAKKDP 32 gi ref NP_ MKWVWALLLLAAW--AAAERDCRVSSFRVKENFDKARFSGTWYAMAKKDP 48 gi sp Q00724 RETB_MOUS MEWVWALVLLAALGGGSAERDCRVSSFRVKENFDKARFSGLWYAIAKKDP 50 gi sp P04916 RETB_RAT MEWVWALVLLAALGGGSAERDCRVSSFRVKENFDKARFSGLWYAIAKKDP 50 ********************:* ***:***** gi pir A39486 EGLFLQDNIVAEFSVDENGHMSATAKGRVRLLNNWDVCADMVGTFTDTED 100 gi sp P18902 RETB_BOVIN EGLFLQDNIVAEFSVDENGHMSATAKGRVRLLNNWDVCADMVGTFTDTED 82 gi ref NP_ EGLFLQDNIVAEFSVDETGQMSATAKGRVRLLNNWDVCADMVGTFTDTED 98 gi sp Q00724 RETB_MOUS EGLFLQDNIIAEFSVDEKGHMSATAKGRVRLLSNWEVCADMVGTFTDTED 100 gi sp P04916 RETB_RAT EGLFLQDNIIAEFSVDEKGHMSATAKGRVRLLSNWEVCADMVGTFTDTED 100 *********:*******.*:************.**:************** gi pir A39486 PAKFKMKYWGVASFLQKGNDDHWIIDTDYDTYAAQYSCRLQNLDGTCADS 150 gi sp P18902 RETB_BOVIN PAKFKMKYWGVASFLQKGNDDHWIIDTDYETFAVQYSCRLLNLDGTCADS 132 gi ref NP_ PAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAVQYSCRLLNLDGTCADS 148 gi sp Q00724 RETB_MOUS PAKFKMKYWGVASFLQRGNDDHWIIDTDYDTFALQYSCRLQNLDGTCADS 150 gi sp P04916 RETB_RAT PAKFKMKYWGVASFLQRGNDDHWIIDTDYDTFALQYSCRLQNLDGTCADS 150 ****************:*******:****:*:* ****** ********* Fig Page 326
32 Progressive MSA stage 3 of 3: progressively align the sequences following the branch order of the tree: Order matters THE LAST FAT CAT THE FAST CAT THE VERY FAST CAT THE FAT CAT THE LAST FAT CAT THE FAST CAT --- THE LAST FA-T CAT THE FAST CA-T --- THE VERY FAST CAT Adapted from C. Notredame, Pharmacogenomics 2002 THE LAST FA-T CAT THE FAST CA-T --- THE VERY FAST CAT THE ---- FA-T CAT
33 Progressive MSA stage 3 of 3: progressively align the sequences following the branch order of the tree: Order matters THE FAT CAT THE FAST CAT THE VERY FAST CAT THE LAST FAT CAT THE FA-T CAT THE FAST CAT THE ---- FA-T CAT THE ---- FAST CAT THE VERY FAST CAT Adapted from C. Notredame, Pharmacogenomics 2002 THE ---- FA-T CAT THE ---- FAST CAT THE VERY FAST CAT THE LAST FA-T CAT
34 Progressive MSA stage 3 of 3: progressively align the sequences following the branch order of the tree: CLUSTALW results
35 MUSCLE: next-generation progressive MSA 1) Build a draft progressive alignment Determine pairwise similarity through k-mer counting (not by alignment) Compute distance (triangular distance) matrix Construct tree using UPGMA Construct draft progressive alignment following tree
36 MUSCLE: next-generation progressive MSA 2) Improve the progressive alignment Compute pairwise identity through current MSA Construct new tree with Kimura distance measures Compare new and old trees: if improved, repeat this step, if not improved, then we re done
37 MUSCLE: next-generation progressive MSA 3) Refinement of the MSA Split tree in half by deleting one edge Make profiles of each half of the tree Re-align the profiles Accept/reject the new alignment
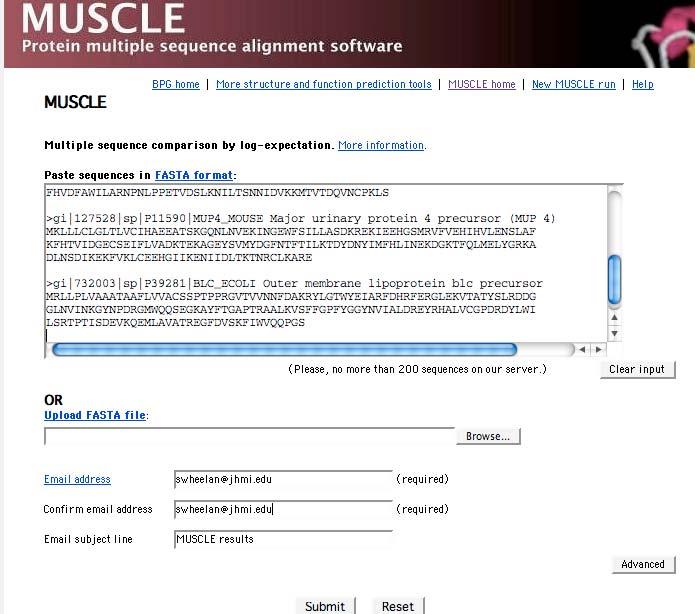
38

39 MUSCLE output (formatted with SeaView)

40 Praline output for the same alignment: pure iterative approach
41 ProbCons consistency-based approach Combines iterative and progressive approaches with a unique probabilistic model. Uses Hidden Markov Models (more in a minute) to calculate probability matrices for matching residues, uses this to construct a guide tree Progressive alignment hierarchically along guide tree Post-processing and iterative refinement (a little like MUSCLE)
42 ProbCons consistency-based approach

43 ProbCons output for the same alignment: how consistency iteration helps
44 Multiple sequence alignment to profile HMMs in 90 s people began to see that aligning sequences to profiles gave much more information than pairwise alignment alone. Hidden Markov models (HMMs) are states that describe the probability of having a particular amino acid residue at arranged In a column of a multiple sequence alignment HMMs are probabilistic models Like a hammer is more refined than a blast, an HMM gives more sensitive alignments than traditional techniques such as progressive alignments Page 325

45 Simple Markov Model Rain = dog may not want to go outside Sun = dog will probably go outside 0.85 S Markov condition = no dependency on anything but nearest previous state ( memoryless ) 0.2 R
46 Simple Hidden Markov Model 0.15 S P(dog goes out in rain) = 0.1 P(dog goes out in sun) = 0.85 R Observation: YNNNYYNNNYN 0.2 (Y=goes out, N=doesn t go out) What is underlying reality (the hidden state chain)?
47 An HMM is constructed from a MSA Example: five lipocalins GTWYA (hs RBP) GLWYA (mus RBP) GRWYE (apod) GTWYE (E Coli) GEWFS (MUP4) Fig Page 327
48 GTWYA GLWYA GRWYE GTWYE GEWFS Prob p(g) 1.0 p(t) 0.4 p(l) 0.2 p(r) 0.2 p(e) p(w) 1.0 p(y) 0.8 p(f) 0.2 p(a) 0.4 p(s) 0.2 Fig Page 327
49 GTWYA GLWYA GRWYE GTWYE GEWFS Prob p(g) 1.0 p(t) 0.4 p(l) 0.2 p(r) 0.2 p(e) p(w) 1.0 p(y) 0.8 p(f) 0.2 p(a) 0.4 p(s) 0.2 P(GEWYE) = (1.0)(0.2)(1.0)(0.8)(0.4) = log odds score = ln(1.0) + ln(0.2) + ln(1.0) + ln(0.8) + ln(0.4) = Fig Page 327
50 GTWYA GLWYA GRWYE GTWYE GEWFS P(GEWYE) = (1.0)(0.2)(1.0)(0.8)(0.4) = log odds score = ln(1.0) + ln(0.2) + ln(1.0) + ln(0.8) + ln(0.4) = G:1.0 T:0.4 L:0.2 R:0.2 E:0.2 W:1.0 Y:0.8 F:0.2 E:0.4 A:0.4 S:0.2 Fig Page 327
51 Structure of a hidden Markov model (HMM) p 4 p 2 I x p 1 M p 3 p 5 p 6 I y p 7
52 Structure of a hidden Markov model (HMM): Trellis representation Fig Page 328
53 HMMER: build a hidden Markov model Determining effective sequence number... done. [4] Weighting sequences heuristically... done. Constructing model architecture... done. Converting counts to probabilities... done. Setting model name, etc.... done. [x] Constructed a profile HMM (length 230) Average score: bits Minimum score: bits Maximum score: bits Std. deviation: bits Fig Page 329
54 HMMER: calibrate a hidden Markov model HMM file: lipocalins.hmm Length distribution mean: 325 Length distribution s.d.: 200 Number of samples: 5000 random seed: histogram(s) saved to: [not saved] POSIX threads: HMM : x mu : lambda : max : Fig Page 329
55 HMMER: search an HMM against GenBank Scores for complete sequences (score includes all domains): Sequence Description Score E-value N gi ref XP_ (XM_129259) ret e gi sp P04916 RETB_RAT Plasma retinol e gi ref XP_ (XM_005907) sim e gi ref NP_ (NM_006744) ret e gi sp P02753 RETB_HUMAN Plasma retinol e gi ref NP_ (NC_003197) out e-90 1 gi ref NP_ : domain 1 of 1, from 1 to 195: score 454.6, E = 1.7e-131 *->mkwvmkllllaalagvfgaaerdafsvgkcrvpspprgfrvkenfdv mkwv++llllaa + +aaerd Crv+s frvkenfd+ gi MKWVWALLLLAA--W--AAAERD------CRVSS----FRVKENFDK 33 erylgtwyeiakkdprferglllqdkitaeysleehgsmsataegrirvl +r++gtwy++akkdp E GL+lqd+I+Ae+S++E+G+Msata+Gr+r+L gi ARFSGTWYAMAKKDP--E-GLFLQDNIVAEFSVDETGQMSATAKGRVRLL 80 enkelcadkvgtvtqiegeasevfltadpaklklkyagvasflqpgfddy +N+++cAD+vGT+t++E dpak+k+ky+gvasflq+g+dd+ gi NNWDVCADMVGTFTDTE DPAKFKMKYWGVASFLQKGNDDH 120 Fig Page 330
56 HMMER: search an HMM against GenBank match to a bacterial lipocalin gi ref NP_ : domain 1 of 1, from 1 to 177: score 318.2, E = 1.9e-90 *->mkwvmkllllaalagvfgaaerdafsvgkcrvpspprgfrvkenfdv M+LL+ +A a ++ Af+v++C++p+PP+G++V++NFD+ gi MRLLPVVA------AVTA-AFLVVACSSPTPPKGVTVVNNFDA 36 erylgtwyeiakkdprferglllqdkitaeysleehgsmsataegrirvl +rylgtwyeia+ D+rFErGL + +ta+ysl++ +G+i+V+ gi KRYLGTWYEIARLDHRFERGL---EQVTATYSLRD DGGINVI 75 enkelcadkvgtvtqiegeasevfltadpaklklkyagvasflqpgfddy Nk++++D EG+a ++t+ P +++lk+ Sf++p++++y gi NKGYNPDR-EMWQKTEGKA---YFTGSPNRAALKV----SFFGPFYGGY 116 Fig Page 330
57 HMMER: search an HMM against GenBank Scores for complete sequences (score includes all domains): Sequence Description Score E-value N gi sp P27485 RETB_PIG Plasma retinol e gi pir A39486 plasma retinol e gi ref XP_ (XM_129259) ret e gi sp P04916 RETB_RAT Plasma retinol e gi ref XP_ (XM_005907) sim e gi sp P02753 RETB_HUMAN Plasma retinol e gi ref NP_ (NM_006744) ret e gi ref NP_ : domain 1 of 1, from 1 to 199: score 600.2, E = 2.6e-175 *->mewvwalvllaalggasaerdcrvssfrvkenfdkarfsgtwyaiak m+wvwal+llaa+ a+aerdcrvssfrvkenfdkarfsgtwya+ak gi MKWVWALLLLAAW--AAAERDCRVSSFRVKENFDKARFSGTWYAMAK 45 KDPEGLFLqDnivAEFsvDEkGhmsAtAKGRvRLLnnWdvCADmvGtFtD KDPEGLFLqDnivAEFsvDE+G+msAtAKGRvRLLnnWdvCADmvGtFtD gi KDPEGLFLQDNIVAEFSVDETGQMSATAKGRVRLLNNWDVCADMVGTFTD 95 tedpakfkmkywgvasflqkgnddhwiidtdydtfavqyscrllnldgtc tedpakfkmkywgvasflqkgnddhwi+dtdydt+avqyscrllnldgtc gi TEDPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAVQYSCRLLNLDGTC 145 Fig Page 330
58 Databases of multiple sequence alignments BLOCKS CDD DOMO INTERPRO iproclass MetaFAM PFAM PRINTS PRODOM PROSITE SMART Page
59 Databases of multiple sequence alignments BLOCKS (HMM) CDD (HMM) DOMO (Gapped MSA) INTERPRO iproclass MetaFAM Pfam (profile HMM library) PRINTS PRODOM (PSI-BLAST) PROSITE SMART Page
60 Databases of multiple sequence alignments BLOCKS CDD DOMO INTERPRO iproclass MetaFAM PFAM PRINTS PRODOM PROSITE SMART Integrative resources Page

61 Pfam (protein family) database: or
")
62 Pfam (protein family) database
")
63 Pfam (protein family) database
")
64 Pfam (protein family) database
65 Pfam (protein family) database
 Search for conserved domains")
66 SMART: Simple Modular Architecture Research Tool (emphasis on cell signaling) Search for conserved domains Page 338
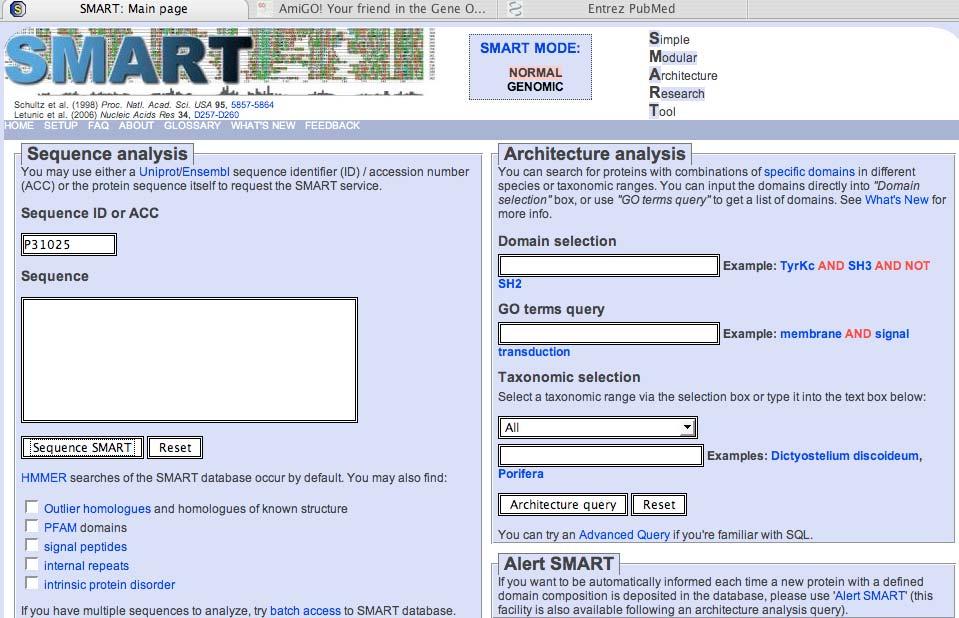
67
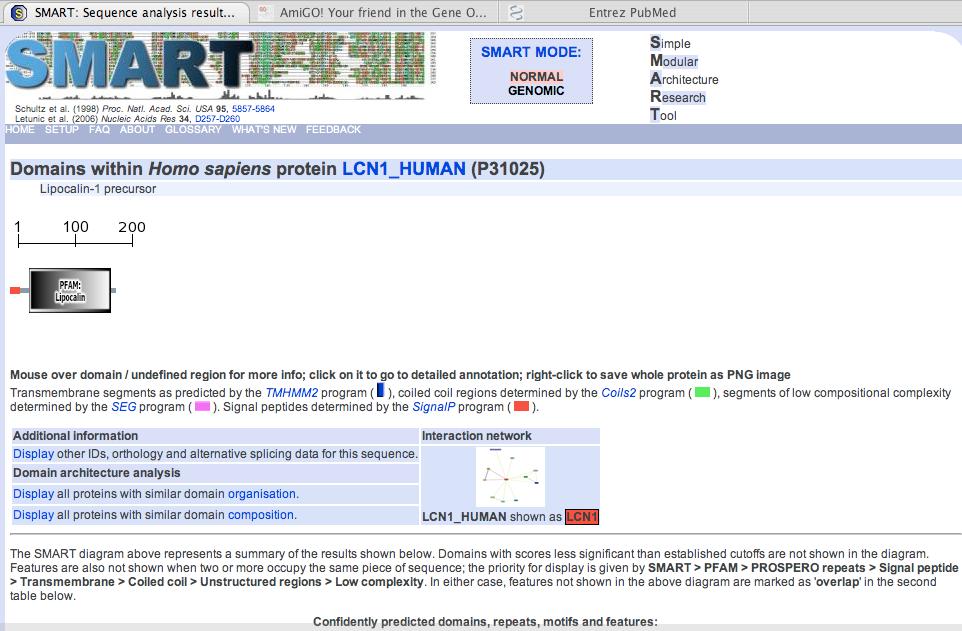
68
69 Databases of multiple sequence alignments BLOCKS CDD DOMO INTERPRO iproclass MetaFAM PFAM PRINTS PRODOM PROSITE SMART CDD at NCBI = PFAM + SMART Page 333
70 CDD uses RPS-BLAST: reverse position-specific Query = your favorite protein Database = set of many PSSMs CDD is related to PSI-BLAST, but distinct CDD searches against profiles generated from pre-selected alignments Purpose: to find conserved domains in the query sequence You can access CDD via DART at NCBI Page 333

71 CDART (from NCBI) allows CDD searches
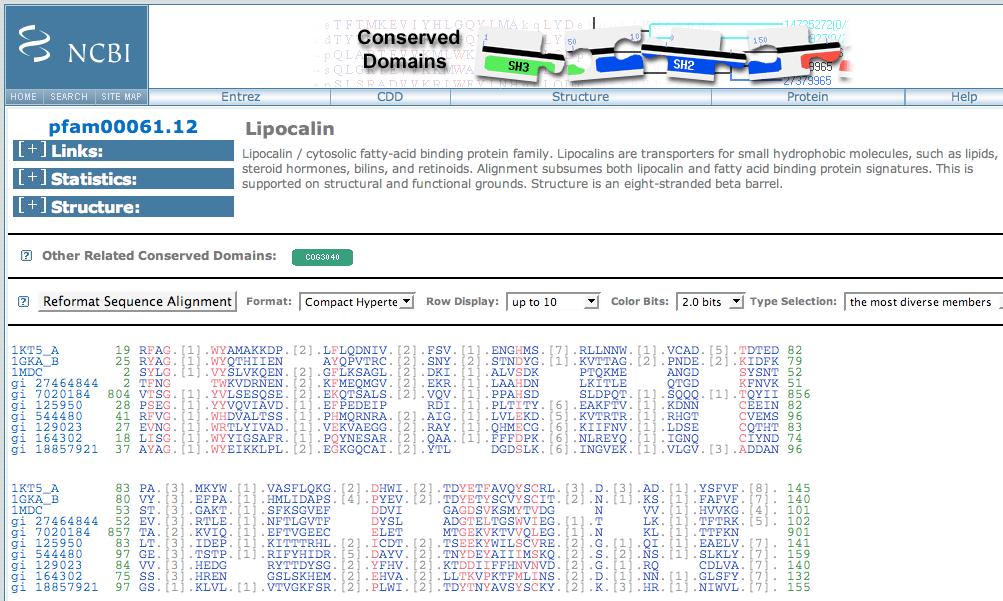
72
73 BLOCKS server BLOCKS: ungapped MSA Very highly conserved regions
74

75
76
77
78
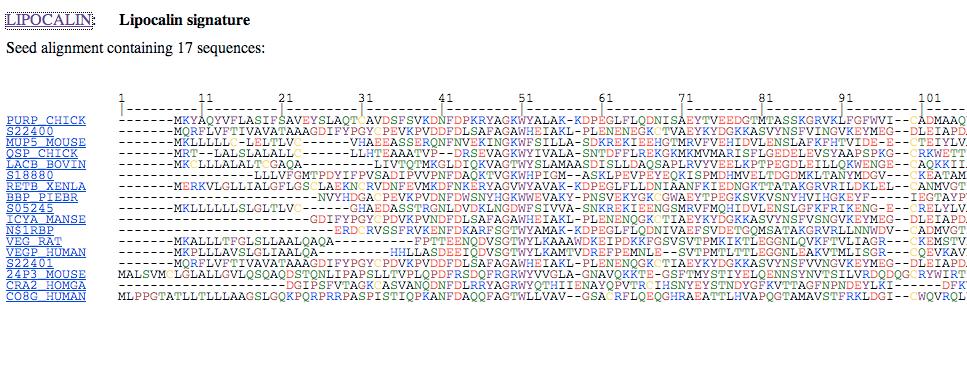
79

80 Fig Page 341
81

82 See chapter 8


83 PROSITE has a syntax to define signatures See chapter 8

84 PopSet: polymorphisms in population data Fig Page 342

85 PopSet: polymorphisms in population data Fig Page 342

86 PopSet: polymorphisms in population data
87 Big Picture sites very useful eview-edital-desc.html Nice overview site lots of alignment utilities, including editors and viewers. Most are current. Pedro s molecular biology tools. Most are current.
88 MSA databases: manual vs. automated curation Manual curation: Pfam PROSITE BLOCKS PRINTS Automated curation: DOMO PRODOM MetaFam + comprehensive - alignment errors
89 Multiple sequence alignment programs AMAS CINEMA ClustalW ClustalX DIALIGN HMMT Match-Box MultAlin MSA Musca PileUp SAGA T-COFFEE
90 Selecting sequences for a PileUp in GCG Fig Page 346
91 GCG PileUp Fig Page 346

92 GCG PileUp Fig Page 347
93 Multiple sequence alignment algorithms Local Global Progressive PIMA CLUSTAL PileUp other Iterative DIALIGN SAGA Fig Page 348
94 Strategy for assessment of alternative multiple sequence alignment algorithms [1] Create or obtain a database of protein sequences for which the 3D structure is known. Thus we can define true homologs using structural criteria. [2] Try making multiple sequence alignments with many different sets of proteins (very related, very distant, few gaps, many gaps, insertions, outliers). [3] Compare the answers. Page 346
95 Strategy for assessment of alternative multiple sequence alignment algorithms Commonly used benchmarking databases: BAliBASE SMART SABmark PREFAB (Protein Reference Alignment Benchmark)
96 Conclusions: assessment of alternative multiple sequence alignment algorithms [1] As percent identity among proteins drops, performance (accuracy) declines also. This is especially severe for proteins < 25% identity. Proteins <25% identity: 65% of residues align well Proteins <40% identity: 80% of residues align well Page 350
97 Conclusions: assessment of alternative multiple sequence alignment algorithms [2] Orphan sequences are highly divergent members of a family. Surprisingly, orphans do not disrupt alignments. Page 350
98 Conclusions: assessment of alternative multiple sequence alignment algorithms [3] Separate multiple sequence alignments can be combined (e.g. RBPs and lactoglobulins). Profile-based methods are very powerful but the strength of the profile depends on the quality of the initial alignment. Page 350
99 Conclusions: assessment of alternative multiple sequence alignment algorithms [4] When proteins have large N-terminal or C-terminal extensions, local alignment algorithms are superior. PileUp (global) is an exception. Page 350
Multiple sequence alignment
 Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
Copyright notice. Multiple sequence alignment. Multiple sequence alignment: outline. Multiple sequence alignment: today s goals
 Copyright notice Multiple sequence alignment Monday, December 8, 2008 Many of the images in this powerpoint presentation are from Bioinformatics and Functional Genomics by J Pevsner (ISBN 0-471-21004-8).
Copyright notice Multiple sequence alignment Monday, December 8, 2008 Many of the images in this powerpoint presentation are from Bioinformatics and Functional Genomics by J Pevsner (ISBN 0-471-21004-8).
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Copyright 2000 N. AYDIN. All rights reserved. 1
 Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Multiple Sequence Alignment: A Critical Comparison of Four Popular Programs
 Multiple Sequence Alignment: A Critical Comparison of Four Popular Programs Shirley Sutton, Biochemistry 218 Final Project, March 14, 2008 Introduction For both the computational biologist and the research
Multiple Sequence Alignment: A Critical Comparison of Four Popular Programs Shirley Sutton, Biochemistry 218 Final Project, March 14, 2008 Introduction For both the computational biologist and the research
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
Sequence Analysis '17- lecture 8. Multiple sequence alignment
 Sequence Analysis '17- lecture 8 Multiple sequence alignment Ex5 explanation How many random database search scores have e-values 10? (Answer: 10!) Why? e-value of x = m*p(s x), where m is the database
Sequence Analysis '17- lecture 8 Multiple sequence alignment Ex5 explanation How many random database search scores have e-values 10? (Answer: 10!) Why? e-value of x = m*p(s x), where m is the database
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT
 5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
InDel 3-5. InDel 8-9. InDel 3-5. InDel 8-9. InDel InDel 8-9
 Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
The PRALINE online server: optimising progressive multiple alignment on the web
 Computational Biology and Chemistry 27 (2003) 511 519 Software Note The PRALINE online server: optimising progressive multiple alignment on the web V.A. Simossis a,b, J. Heringa a, a Bioinformatics Unit,
Computational Biology and Chemistry 27 (2003) 511 519 Software Note The PRALINE online server: optimising progressive multiple alignment on the web V.A. Simossis a,b, J. Heringa a, a Bioinformatics Unit,
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Introductory course on Multiple Sequence Alignment Part I: Theoretical foundations
 Sequence Analysis and Structure Prediction Service Centro Nacional de Biotecnología CSIC 8-10 May, 2013 Introductory course on Multiple Sequence Alignment Part I: Theoretical foundations Course Notes Instructor:
Sequence Analysis and Structure Prediction Service Centro Nacional de Biotecnología CSIC 8-10 May, 2013 Introductory course on Multiple Sequence Alignment Part I: Theoretical foundations Course Notes Instructor:
Introduction to Bioinformatics Online Course: IBT
 Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
CONCEPT OF SEQUENCE COMPARISON. Natapol Pornputtapong 18 January 2018
 CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
Protein Bioinformatics. Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet sandberg.cmb.ki.
 Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
Motifs, Profiles and Domains. Michael Tress Protein Design Group Centro Nacional de Biotecnología, CSIC
 Motifs, Profiles and Domains Michael Tress Protein Design Group Centro Nacional de Biotecnología, CSIC Comparing Two Proteins Sequence Alignment Determining the pattern of evolution and identifying conserved
Motifs, Profiles and Domains Michael Tress Protein Design Group Centro Nacional de Biotecnología, CSIC Comparing Two Proteins Sequence Alignment Determining the pattern of evolution and identifying conserved
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Bioinformatics. Proteins II. - Pattern, Profile, & Structure Database Searching. Robert Latek, Ph.D. Bioinformatics, Biocomputing
 Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Some Problems from Enzyme Families
 Some Problems from Enzyme Families Greg Butler Department of Computer Science Concordia University, Montreal www.cs.concordia.ca/~faculty/gregb gregb@cs.concordia.ca Abstract I will discuss some problems
Some Problems from Enzyme Families Greg Butler Department of Computer Science Concordia University, Montreal www.cs.concordia.ca/~faculty/gregb gregb@cs.concordia.ca Abstract I will discuss some problems
Sara C. Madeira. Universidade da Beira Interior. (Thanks to Ana Teresa Freitas, IST for useful resources on this subject)
") Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Similarity searching summary (2)
") Similarity searching / sequence alignment summary Biol4230 Thurs, February 22, 2016 Bill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 What have we covered? Homology excess similiarity but no excess similarity
Similarity searching / sequence alignment summary Biol4230 Thurs, February 22, 2016 Bill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 What have we covered? Homology excess similiarity but no excess similarity
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
A profile-based protein sequence alignment algorithm for a domain clustering database
 A profile-based protein sequence alignment algorithm for a domain clustering database Lin Xu,2 Fa Zhang and Zhiyong Liu 3, Key Laboratory of Computer System and architecture, the Institute of Computing
A profile-based protein sequence alignment algorithm for a domain clustering database Lin Xu,2 Fa Zhang and Zhiyong Liu 3, Key Laboratory of Computer System and architecture, the Institute of Computing
Bioinformatics. Dept. of Computational Biology & Bioinformatics
 Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Christian Sigrist. November 14 Protein Bioinformatics: Sequence-Structure-Function 2018 Basel
 Christian Sigrist General Definition on Conserved Regions Conserved regions in proteins can be classified into 5 different groups: Domains: specific combination of secondary structures organized into a
Christian Sigrist General Definition on Conserved Regions Conserved regions in proteins can be classified into 5 different groups: Domains: specific combination of secondary structures organized into a
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Alignment principles and homology searching using (PSI-)BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)
BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)") Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Probalign: Multiple sequence alignment using partition function posterior probabilities
 Sequence Analysis Probalign: Multiple sequence alignment using partition function posterior probabilities Usman Roshan 1* and Dennis R. Livesay 2 1 Department of Computer Science, New Jersey Institute
Sequence Analysis Probalign: Multiple sequence alignment using partition function posterior probabilities Usman Roshan 1* and Dennis R. Livesay 2 1 Department of Computer Science, New Jersey Institute
Multiple sequence alignments
 Multiple sequence alignments Special thanks to all the scientis that made public available their presentations throughout the web from where many slides were taken to eleborate this presentation Web sites
Multiple sequence alignments Special thanks to all the scientis that made public available their presentations throughout the web from where many slides were taken to eleborate this presentation Web sites
Chapter 11 Multiple sequence alignment
 Chapter 11 Multiple sequence alignment Burkhard Morgenstern 1. INTRODUCTION Sequence alignment is of crucial importance for all aspects of biological sequence analysis. Virtually all methods of nucleic
Chapter 11 Multiple sequence alignment Burkhard Morgenstern 1. INTRODUCTION Sequence alignment is of crucial importance for all aspects of biological sequence analysis. Virtually all methods of nucleic
Multiple Alignment. Anders Gorm Pedersen Molecular Evolution Group Center for Biological Sequence Analysis
 Multiple Alignment Anders Gorm Pedersen Molecular Evolution Group Center for Biological Sequence Analysis gorm@cbs.dtu.dk Refresher: pairwise alignments 43.2% identity; Global alignment score: 374 10 20
Multiple Alignment Anders Gorm Pedersen Molecular Evolution Group Center for Biological Sequence Analysis gorm@cbs.dtu.dk Refresher: pairwise alignments 43.2% identity; Global alignment score: 374 10 20
Sequence Analysis and Databases 2: Sequences and Multiple Alignments
 1 Sequence Analysis and Databases 2: Sequences and Multiple Alignments Jose María González-Izarzugaza Martínez CNIO Spanish National Cancer Research Centre (jmgonzalez@cnio.es) 2 Sequence Comparisons:
1 Sequence Analysis and Databases 2: Sequences and Multiple Alignments Jose María González-Izarzugaza Martínez CNIO Spanish National Cancer Research Centre (jmgonzalez@cnio.es) 2 Sequence Comparisons:
Sequence Bioinformatics. Multiple Sequence Alignment Waqas Nasir
 Sequence Bioinformatics Multiple Sequence Alignment Waqas Nasir 2010-11-12 Multiple Sequence Alignment One amino acid plays coy; a pair of homologous sequences whisper; many aligned sequences shout out
Sequence Bioinformatics Multiple Sequence Alignment Waqas Nasir 2010-11-12 Multiple Sequence Alignment One amino acid plays coy; a pair of homologous sequences whisper; many aligned sequences shout out
Alignment & BLAST. By: Hadi Mozafari KUMS
 Alignment & BLAST By: Hadi Mozafari KUMS SIMILARITY - ALIGNMENT Comparison of primary DNA or protein sequences to other primary or secondary sequences Expecting that the function of the similar sequence
Alignment & BLAST By: Hadi Mozafari KUMS SIMILARITY - ALIGNMENT Comparison of primary DNA or protein sequences to other primary or secondary sequences Expecting that the function of the similar sequence
Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics
 Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics Tandy Warnow Founder Professor of Engineering The University of Illinois at Urbana-Champaign http://tandy.cs.illinois.edu
Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics Tandy Warnow Founder Professor of Engineering The University of Illinois at Urbana-Champaign http://tandy.cs.illinois.edu
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Sequence Analysis 17: lecture 5. Substitution matrices Multiple sequence alignment
 Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Overview Multiple Sequence Alignment
 Overview Multiple Sequence Alignment Inge Jonassen Bioinformatics group Dept. of Informatics, UoB Inge.Jonassen@ii.uib.no Definition/examples Use of alignments The alignment problem scoring alignments
Overview Multiple Sequence Alignment Inge Jonassen Bioinformatics group Dept. of Informatics, UoB Inge.Jonassen@ii.uib.no Definition/examples Use of alignments The alignment problem scoring alignments
Amino Acid Structures from Klug & Cummings. 10/7/2003 CAP/CGS 5991: Lecture 7 1
 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 1 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 2 Amino Acid Structures from Klug & Cummings
Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 1 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 2 Amino Acid Structures from Klug & Cummings
Genomics and bioinformatics summary. Finding genes -- computer searches
 Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Research Proposal. Title: Multiple Sequence Alignment used to investigate the co-evolving positions in OxyR Protein family.
 Research Proposal Title: Multiple Sequence Alignment used to investigate the co-evolving positions in OxyR Protein family. Name: Minjal Pancholi Howard University Washington, DC. June 19, 2009 Research
Research Proposal Title: Multiple Sequence Alignment used to investigate the co-evolving positions in OxyR Protein family. Name: Minjal Pancholi Howard University Washington, DC. June 19, 2009 Research
08/21/2017 BLAST. Multiple Sequence Alignments: Clustal Omega
 BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
BLAST. Varieties of BLAST
 BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I)
 Sequence pairwise alignment (I)") CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
HMM applications. Applications of HMMs. Gene finding with HMMs. Using the gene finder
 HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
Hidden Markov Models (HMMs) and Profiles
 and Profiles") Hidden Markov Models (HMMs) and Profiles Swiss Institute of Bioinformatics (SIB) 26-30 November 2001 Markov Chain Models A Markov Chain Model is a succession of states S i (i = 0, 1,...) connected by transitions.
Hidden Markov Models (HMMs) and Profiles Swiss Institute of Bioinformatics (SIB) 26-30 November 2001 Markov Chain Models A Markov Chain Model is a succession of states S i (i = 0, 1,...) connected by transitions.
Whole Genome Alignments and Synteny Maps
 Whole Genome Alignments and Synteny Maps IINTRODUCTION It was not until closely related organism genomes have been sequenced that people start to think about aligning genomes and chromosomes instead of
Whole Genome Alignments and Synteny Maps IINTRODUCTION It was not until closely related organism genomes have been sequenced that people start to think about aligning genomes and chromosomes instead of
2MHR. Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity.
 Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Heuristic Alignment and Searching
 3/28/2012 Types of alignments Global Alignment Each letter of each sequence is aligned to a letter or a gap (e.g., Needleman-Wunsch). Local Alignment An optimal pair of subsequences is taken from the two
3/28/2012 Types of alignments Global Alignment Each letter of each sequence is aligned to a letter or a gap (e.g., Needleman-Wunsch). Local Alignment An optimal pair of subsequences is taken from the two
Bioinformatics and BLAST
 Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment
 Pairwise sequence alignment") Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Ch. 9 Multiple Sequence Alignment (MSA)
") Ch. 9 Multiple Sequence Alignment (MSA) - gather seqs. to make MSA - doing MSA with ClustalW - doing MSA with Tcoffee - comparing seqs. that cannot align Introduction - from pairwise alignment to MSA -
Ch. 9 Multiple Sequence Alignment (MSA) - gather seqs. to make MSA - doing MSA with ClustalW - doing MSA with Tcoffee - comparing seqs. that cannot align Introduction - from pairwise alignment to MSA -
Bioinformatics 1--lectures 15, 16. Markov chains Hidden Markov models Profile HMMs
 Bioinformatics 1--lectures 15, 16 Markov chains Hidden Markov models Profile HMMs target sequence database input to database search results are sequence family pseudocounts or background-weighted pseudocounts
Bioinformatics 1--lectures 15, 16 Markov chains Hidden Markov models Profile HMMs target sequence database input to database search results are sequence family pseudocounts or background-weighted pseudocounts
A bioinformatics approach to the structural and functional analysis of the glycogen phosphorylase protein family
 A bioinformatics approach to the structural and functional analysis of the glycogen phosphorylase protein family Jieming Shen 1,2 and Hugh B. Nicholas, Jr. 3 1 Bioengineering and Bioinformatics Summer
A bioinformatics approach to the structural and functional analysis of the glycogen phosphorylase protein family Jieming Shen 1,2 and Hugh B. Nicholas, Jr. 3 1 Bioengineering and Bioinformatics Summer
Protein function prediction based on sequence analysis
 Performing sequence searches Post-Blast analysis, Using profiles and pattern-matching Protein function prediction based on sequence analysis Slides from a lecture on MOL204 - Applied Bioinformatics 18-Oct-2005
Performing sequence searches Post-Blast analysis, Using profiles and pattern-matching Protein function prediction based on sequence analysis Slides from a lecture on MOL204 - Applied Bioinformatics 18-Oct-2005
Sequence Analysis, '18 -- lecture 9. Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene.
 Sequence Analysis, '18 -- lecture 9 Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene. How can I represent thousands of homolog sequences in a compact
Sequence Analysis, '18 -- lecture 9 Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene. How can I represent thousands of homolog sequences in a compact
Computational Biology
 Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Sequence Alignments. Dynamic programming approaches, scoring, and significance. Lucy Skrabanek ICB, WMC January 31, 2013
 Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
G4120: Introduction to Computational Biology
 ICB Fall 2003 G4120: Introduction to Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2003 Oliver Jovanovic, All Rights Reserved. Bioinformatics and
ICB Fall 2003 G4120: Introduction to Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2003 Oliver Jovanovic, All Rights Reserved. Bioinformatics and
09/06/25. Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Non-uniform distribution of folds. Scheme of protein structure predicition
 Non-uniform distribution of folds. Scheme of protein structure predicition") Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences
 Analysis of Biological Sequences") First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program)
") Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools. Giri Narasimhan
 CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs15.html Describing & Modeling Patterns
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs15.html Describing & Modeling Patterns
Introduction to protein alignments
 Introduction to protein alignments Comparative Analysis of Proteins Experimental evidence from one or more proteins can be used to infer function of related protein(s). Gene A Gene X Protein A compare
Introduction to protein alignments Comparative Analysis of Proteins Experimental evidence from one or more proteins can be used to infer function of related protein(s). Gene A Gene X Protein A compare
CS612 - Algorithms in Bioinformatics
 Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
Similarity or Identity? When are molecules similar?
 Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Introduction to Bioinformatics
 Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Effects of Gap Open and Gap Extension Penalties
 Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Multiple Sequence Alignment, Gunnar Klau, December 9, 2005, 17:
 Multiple Sequence Alignment, Gunnar Klau, December 9, 2005, 17:50 5001 5 Multiple Sequence Alignment The first part of this exposition is based on the following sources, which are recommended reading:
Multiple Sequence Alignment, Gunnar Klau, December 9, 2005, 17:50 5001 5 Multiple Sequence Alignment The first part of this exposition is based on the following sources, which are recommended reading:
Hidden Markov Models
 Hidden Markov Models Outline CG-islands The Fair Bet Casino Hidden Markov Model Decoding Algorithm Forward-Backward Algorithm Profile HMMs HMM Parameter Estimation Viterbi training Baum-Welch algorithm
Hidden Markov Models Outline CG-islands The Fair Bet Casino Hidden Markov Model Decoding Algorithm Forward-Backward Algorithm Profile HMMs HMM Parameter Estimation Viterbi training Baum-Welch algorithm
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Bioinformatics Definitions The use of computational
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Bioinformatics Definitions The use of computational
Using Bioinformatics to Study Evolutionary Relationships Instructions
 3 Using Bioinformatics to Study Evolutionary Relationships Instructions Student Researcher Background: Making and Using Multiple Sequence Alignments One of the primary tasks of genetic researchers is comparing
3 Using Bioinformatics to Study Evolutionary Relationships Instructions Student Researcher Background: Making and Using Multiple Sequence Alignments One of the primary tasks of genetic researchers is comparing
Exercise 5. Sequence Profiles & BLAST
 Exercise 5 Sequence Profiles & BLAST 1 Substitution Matrix (BLOSUM62) Likelihood to substitute one amino acid with another Figure taken from https://en.wikipedia.org/wiki/blosum 2 Substitution Matrix (BLOSUM62)
Exercise 5 Sequence Profiles & BLAST 1 Substitution Matrix (BLOSUM62) Likelihood to substitute one amino acid with another Figure taken from https://en.wikipedia.org/wiki/blosum 2 Substitution Matrix (BLOSUM62)
BIOINFORMATICS ORIGINAL PAPER doi: /bioinformatics/btm017
 Vol. 23 no. 7 2007, pages 802 808 BIOINFORMATICS ORIGINAL PAPER doi:10.1093/bioinformatics/btm017 Sequence analysis PROMALS: towards accurate multiple sequence alignments of distantly related proteins
Vol. 23 no. 7 2007, pages 802 808 BIOINFORMATICS ORIGINAL PAPER doi:10.1093/bioinformatics/btm017 Sequence analysis PROMALS: towards accurate multiple sequence alignments of distantly related proteins
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Phylogenies Scores for Exhaustive Maximum Likelihood and Parsimony Scores Searches
 Int. J. Bioinformatics Research and Applications, Vol. x, No. x, xxxx Phylogenies Scores for Exhaustive Maximum Likelihood and s Searches Hyrum D. Carroll, Perry G. Ridge, Mark J. Clement, Quinn O. Snell
Int. J. Bioinformatics Research and Applications, Vol. x, No. x, xxxx Phylogenies Scores for Exhaustive Maximum Likelihood and s Searches Hyrum D. Carroll, Perry G. Ridge, Mark J. Clement, Quinn O. Snell
An Introduction to Bioinformatics Algorithms Hidden Markov Models
 Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
SUPPLEMENTARY INFORMATION
 Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Biology Tutorial. Aarti Balasubramani Anusha Bharadwaj Massa Shoura Stefan Giovan
 Biology Tutorial Aarti Balasubramani Anusha Bharadwaj Massa Shoura Stefan Giovan Viruses A T4 bacteriophage injecting DNA into a cell. Influenza A virus Electron micrograph of HIV. Cone-shaped cores are
Biology Tutorial Aarti Balasubramani Anusha Bharadwaj Massa Shoura Stefan Giovan Viruses A T4 bacteriophage injecting DNA into a cell. Influenza A virus Electron micrograph of HIV. Cone-shaped cores are
Moreover, the circular logic
 Moreover, the circular logic How do we know what is the right distance without a good alignment? And how do we construct a good alignment without knowing what substitutions were made previously? ATGCGT--GCAAGT
Moreover, the circular logic How do we know what is the right distance without a good alignment? And how do we construct a good alignment without knowing what substitutions were made previously? ATGCGT--GCAAGT
Upcoming challenges for multiple sequence alignment methods in the high-throughput era
 BIOINFORMATICS REVIEW Vol. 25 no. 19 2009, pages 2455 2465 doi:10.1093/bioinformatics/btp452 Sequence analysis Upcoming challenges for multiple sequence alignment methods in the high-throughput era Carsten
BIOINFORMATICS REVIEW Vol. 25 no. 19 2009, pages 2455 2465 doi:10.1093/bioinformatics/btp452 Sequence analysis Upcoming challenges for multiple sequence alignment methods in the high-throughput era Carsten
Motivating the need for optimal sequence alignments...
 1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT
 3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
Comparing whole genomes
 BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
PROTEIN CLUSTERING AND CLASSIFICATION
 PROTEIN CLUSTERING AND CLASSIFICATION ori Sasson 1 and Michal Linial 2 1The School of Computer Science and Engeeniring and 2 The Life Science Institute, The Hebrew University of Jerusalem, Israel 1. Introduction
PROTEIN CLUSTERING AND CLASSIFICATION ori Sasson 1 and Michal Linial 2 1The School of Computer Science and Engeeniring and 2 The Life Science Institute, The Hebrew University of Jerusalem, Israel 1. Introduction
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Citation BMC Bioinformatics, 2016, v. 17 n. suppl. 8, p. 285:
 Title PnpProbs: A better multiple sequence alignment tool by better handling of guide trees Author(s) YE, Y; Lam, TW; Ting, HF Citation BMC Bioinformatics, 2016, v. 17 n. suppl. 8, p. 285:633-643 Issued
Title PnpProbs: A better multiple sequence alignment tool by better handling of guide trees Author(s) YE, Y; Lam, TW; Ting, HF Citation BMC Bioinformatics, 2016, v. 17 n. suppl. 8, p. 285:633-643 Issued
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
SUPPLEMENTARY INFORMATION
 Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Amino Acid Structures from Klug & Cummings. Bioinformatics (Lec 12)
") Amino Acid Structures from Klug & Cummings 2/17/05 1 Amino Acid Structures from Klug & Cummings 2/17/05 2 Amino Acid Structures from Klug & Cummings 2/17/05 3 Amino Acid Structures from Klug & Cummings
Amino Acid Structures from Klug & Cummings 2/17/05 1 Amino Acid Structures from Klug & Cummings 2/17/05 2 Amino Acid Structures from Klug & Cummings 2/17/05 3 Amino Acid Structures from Klug & Cummings
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5
 Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
Single alignment: Substitution Matrix. 16 march 2017
 Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/8/07 CAP5510 1 Pattern Discovery 2/8/07 CAP5510 2 Patterns Nature
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/8/07 CAP5510 1 Pattern Discovery 2/8/07 CAP5510 2 Patterns Nature
Objectives. Comparison and Analysis of Heat Shock Proteins in Organisms of the Kingdom Viridiplantae. Emily Germain 1,2 Mentor Dr.
 Comparison and Analysis of Heat Shock Proteins in Organisms of the Kingdom Viridiplantae Emily Germain 1,2 Mentor Dr. Hugh Nicholas 3 1 Bioengineering & Bioinformatics Summer Institute, Department of Computational
Comparison and Analysis of Heat Shock Proteins in Organisms of the Kingdom Viridiplantae Emily Germain 1,2 Mentor Dr. Hugh Nicholas 3 1 Bioengineering & Bioinformatics Summer Institute, Department of Computational