Template-Based 3D Structure Prediction
|
|
|
- Elijah Wilkinson
- 6 years ago
- Views:
Transcription
1 Template-Based 3D Structure Prediction Sequence and Structure-based Template Detection and Alignment Issues


2 The rate of new sequences is growing exponentially relative to the rate of protein structures being solved!




3 Why Such a shift? Sequencing DNA is easy= 1-2 days Experimental determination of a protein is difficult= 1-3 years Small targets
4 How could we fill the gap between the number of known sequences and known structures? Structural Genomics Initiatives: JCSG
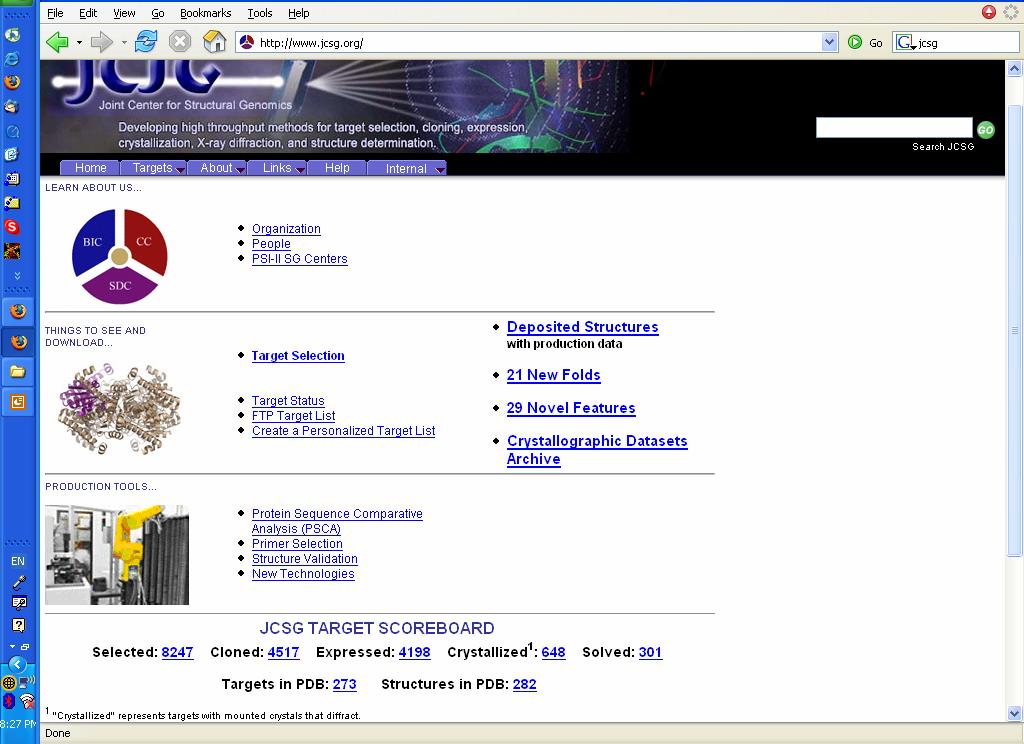
5 2005
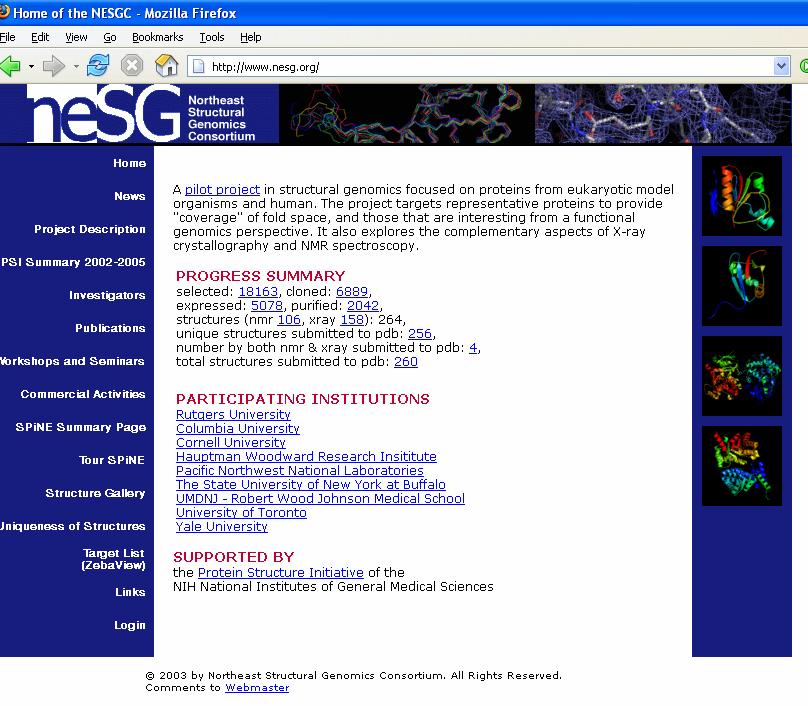
6 2005

7 How could we fill the gap between the number of known sequences and known structures? Structural Genomics Initiative: JCSG or
8 SHORT REMINDER 1D: SECONDARY STRUCTURE ELEMENTS HELIX SHEET LOOPS* 3D=> FOLDING OF THESE SECONDARY STRUCTURE ELEMENTS (SEQUENTIAL and SPATIAL ARRAGEMENT OF SECONDARY STRUCTURE ELEMENTS)


9 Current methods to predict protein structure Structural level Schema Additional info Ab Initio Secondary D 2D 3D 4D AAVLYFGREDHTLLVY 2 nd pred correlated mutations AAVLYFGREDHTLLVY AAVLYFGREDHTLLVY Tertiary Quaternary -molecular dynamics -Energy minimization -docking No Ab-Initio 2 nd pred. -homology modeling -threading -filtered docking



10 3D?? MREYKLVVLGSGGVGKSALTVQFVQGIFVDE YDPTIEDSYRKQVEVDCQQCMLEILDTAGTE QFTAMRDLYMKNGQGFALVYSITAQSTFNDL QDLREQILRVKDTEDVPMILVGNKCDLEDER VVGKEQGQNLARQWCNCAFLESSAKSKINVN EIFYDLVRQINR? How does it fold?"
11 So How Do You Get from Query Sequence to Model Structure? Template Detection The first step is to find a sufficiently similar structural template or templates from the PDB, either by sequence searches or more sophisticated structure-based techniques. Alignment All template detection methods need to create alignments in order to be able to evaluate the query-template fit. Alignments are also crucial for the next stage... Model Building Ranges from the simple tranference of PDB coordinates built into many fold recognition methods to complex all atom compative modelling. Evaluation All methods again use some sort of quality assessment of the models, either at the level of the alignment or of the feasibility of the 3D structure.

12 Template Identification Template Detection The most simple form of template detection is a sequence search of the sequences in the PDB database. This should always be the first step because the results of this search will condition the approach. Domains Searching for templates is complicated by the fact that many proteins are made up of several structural domains. A domain search should be carried out at the same time as the sequence search.
13 Russell: Structural prediction flowchart

14 Template Detection Continued But, if No Similar PDB Template Exists If no template is found for one or more of the domains, more work will be needed, particularly with the alignment, in order to produce a good model. In this case the predictor can move onto more complex sequence search methods (PSIBLAST, FFAS, HMMs) or use fold recognition techniques.

15 Structural prediction flowchart
16 Homology Modelling vs Fold Recognition % seq. ID Application Fold Recognition Homology Modelling Target Sequence Model Quality Any Sequence Fold Level >= 30-50% ID with template Atomic Level If the sequence is similar to a known structure (>30-50% identity) you can usually move straight onto generating an all atom model by homology modelling.
17 No Template Found by BLAST? Pairwise sequence search methods can detect folds when sequence similarity is high,. but are very poor at detecting relationships that have less than 20% identity. One possibility is to use profile-based sequence search methods. These have evolved greatly, and can find templates with very low sequence similarity. Fold recognition methods can find folds that are too distantly related to be detected by sequence based methods, because they evaluate not only sequence similarity, but also structural fit.
18 Why We Can Build Structures? Because Small Changes in Sequence Have Little Effect on Structure


19 Relationship between sequence and structural similarity Chotia & Lesk, 1986 %id seq. => same 3D (for sure) %id seq. => sometimes same str. sometimes not} depends on the length of the aligned region.

20 Sequence Space vs. Structure Space Homology Modelling Targets Fold Recognition Targets Sequence space Structural space The development of fold recognition methods came from the observation that many apparently unrelated sequences had very similar 3-dimensional structures (folds).
21 FOLD RECOGNITION Find out the real structure with prediction methods FIT SEQUENCES INTO STRUCTURES AND FIND THE BEST MATCH when? If Little Sequence Similarity Then, Fold Recognition
22 FOLD RECOGNITION BIOLOGIST s APPROACH: If seq 1 is similar to seq2 then structure 1 is similar to structure2 and there is probably an evolutionary explanation! PHYSICIST s APPROACH: Proteins form structures according to fundamental rules that they call energies or free energies! Quoted from: Protein Structure Prediction, Huber & Torda.s
23 Fold Recognition Algorithms: General Principle It was thought when fold recognition methods were developed that they could detect analogues, proteins that were structurally similar but that had no evolutionary relationship. In fact most of these predictions were later shown to be homologous (have an evolutionary relationship) by advanced sequence comparison methods, such as PSI-BLAST. They still have a place though, in part because many of the newer methods are more more sensitive than PSIBLAST, in part because research also shows that no one method can always hope to correctly identify a fold.
24 CAPABLE TO DETECT VERY DISTANT HOMOLOGY (WHEN SEQUENCE-BASED METHODS FAIL) FFAS03 example
25 FOLD RECOGNITION FOLD DETECTION THREADING BLAST, FASTA eg. FFAS03 GenThreader FOLD RECOGNITION eg HMM Alignment of sequences to structures as in THREADER (Jones et al. 1992) CAPABLE TO DETECT VERY DISTANT HOMOLOGY (WHEN SEQUENCE-BASED METHODS FAIL) Fold recognition: distant/no clear homology
26 FOLD RECOGNITION WHAT IS THREADING? To fit a structure into a sequence!..given a protein structure, what amino acid sequences are likely to fold into that structure?
27 QUERY TO STRUCTURE ALIGNMENT S1 S2 S3 S4 S5 Sheet helix Optimal alignments Suboptimal alignments
28 QUERY TO STRUCTURE ALIGNMENT I query sequence Structure template ALIGNMENT (threading): covering of segments of the query sequence by template blocks! A threading is completely determined by the starting positions of the blocks
29 QUERY TO STRUCTURE ALIGNMENT II: Rules query sequence The blocks preserve their order Structure template The blocks DO NOT OVERLAP There is NO GAPS in the blocks!
30 STEPS Construct a library of Potential core folds (structural templates) Choose an objective function (score function) to evaluate any alignment of a sequence to a structure template
31 The General Principle I 1. Library of protein structures (fold library) all known structures representative subset (seq. similarity filters) structural cores with loops removed
32 Building a Fold library
33 The General Principle II 2. Binary alignment algorithm with Scoring function contact potential environments Others.. Instead of aligning a sequence to a sequence, align strings of descriptors that represent 3D structural features. Usual Dynamic Programming: score matrix relates two amino acids Threading Dynamic Programming: relates amino acids to environments in 3D structure ALMVWTGH Evaluation of the fitness: probability The final score is the goodness of fit of the target sequence to each fold and is usually reported as a probability....
34 Position j=4 j=3 j=2 j=1 S T i=1 i=2 i=3 i=4 i=5 i=6 Block Each possible threading corresponds to a path from S to T in the graph and vice-versa The BLUE path corresponds to the threading (1,4,1,4,1,4) The GREEN path corresponds to the threading (1,2,2,3,4,4) THE KEY IS TO FIND THE SHORTEST PATH FROM S TO T =dynamic programming!!!
35 Scoring Functions for Fold Recognition Scoring functions measure some or more of the following: The similarity between the observed structural environment of the residue and the environment in which the residue is usually found Pair potentials Solvation energy Coincidence of real and predicted secondary structure and accessibility Evolutionary information (from aligned structures and sequences)
36 Structural Environments Bowie et al. (1991) created a fold recognition approach: each position of a fold template as being in one of eighteen environments. Environment: measuring the side chain buried area, the fraction of the side chain area that was exposed to polar atoms, and the local secondary structure. Other researches have developed similar methods, where the structural environments described include exposed atomic areas and type of residue-residue contacts.
37 How Structural Environments Scores are Used 20aa 18 env i.e.: Prob to have K Buried. Scoring matrices are pre-generated for the probabilities of finding each of the twenty amino acids in each of the environment classes. Probabilities are drawn from databases of known structures. Using these probabilities a 3D profile is created for each fold in the fold library. #This 3D matrix defines the probability of finding a certain amino acid in a certain position in each fold. When the target sequence is aligned with the fold, a score is calculated from the pre-generated 3D profile for each of the positions in the alignment. The fit of a fold is the sum of the probabilities of each residue being found in each environment.
38 Solvation Energy Solvation potential is a term used to describe the preference of an amino acid for a specific level of residue burial. It is derived by comparing the frequency of occurrence of each amino acid at a specific degree of residue burial to the frequency of occurrence of all other amino acid types with this degree of burial. The degree of burial of a residue is defined as the ratio between its solvent accessible surface area and its overall surface area.
39 Pair or Contact Potentials - the Tendency of residues to be in Contact counts d Counts become propensities (frequency at each distance separation) or energies (Boltzmann principle, -KT ln) Make count of interacting pairs of each residue type at different distance separations E d
40 Pair Potentials in Fold Recognition The energy that results from aligning a certain target sequence residue at a certain position depends on its interactions with other residues. This creates problems when pair potentials are used to create sequence structure alignments, since you do not know the position of all the residues in the model before threading them. Threading methods that use pair potentials in this way, such as THREADER (Jones et al, 1992) have to use clever programming methods to get round this problem.




41 INPUT Secondary structure pred TOPITS uses predicted secondary structure and accessibilities for the target sequence and compares them with the known values of the template. Rost, 1995
42 Alignments 1aac DKATIPSEPFAAAEVADGAIVVDIAKMKYETPELHVKVGDTVTWINREAMPHNVHFVAGV :.... :... :.... :..:::. : 1plc IDVLLGADDGSLAFVPSEFSISPGEKIVFKNNAGFPHNIVFDEDS 1aac L--GEAALKGPMMKKE------QAYSLTFTEAGTYDYHCTPHPF--MRGKVVVE. : : : : : :...:.:: : :::.:. 1plc IPSGVDASKISMSEEDLLNAKGETFEVALSNKGEYSFYCSPHQGAGMVGKVTVN All methods of template detection, whether sequence-based, fold recognition or hybrid needs alignments between the query sequence and the PDB template sequence. The quality of these alignments is highly variable. If an accurate 3D model is to be built, it is vital that the target-template alignments are correct. Particularly at lower percentage identity the biggest errors stem from the alignments.

43 Alignments Generally the higher the sequence similarity and the lower the number of gaps between the two sequences, the more likely the alignment is to be correct. The more sequences that are included in the alignment the more likely the alignment is to be reliable in an evolutionary sense. Coincidence of real and predicted secondary structure and accessibility also generally improves alignments. Even with all this information automatic methods are far from perfect.
44 Alignments by Hand Alignments from sequence-based methods tend to produce alignments that are biased towards sequence evolution not structure and fold recognition alignments are not any more reliable. In practice most predictors update alignments manually using actual and predicted secondary structure and accessibility information, and careful placement of gaps. KSLKGSRTEKNILTAFAGESQARNRYNYFGGQAKKDGFVQISDIFAETADQEREHAKRLFKFLE GGDLEIVAAFPAGI. ::---========+==++==+====-==--::-======+==++=++==+====== MKGDTKVINYLNKLLGNELVAINQYFLHARMFKNWGLKRLNDVEYHESIDEMKHADRYIERILFLEGLPNLQDLGKLNI IADTHANLIASAAGEHHEYTEMYPSFARIAREEGYEEIARVFASIAVAEEFHEKRFLDFARNIKE GRVFLREQATK.:---===-=+--==--=- --==-==------:--======-====++==+====----:-:::.. GEDVEEMLRSDLALELDGA KNLREAIGYADSVHDYVSRDMMIEILRDEEGHIDWLETELDLIQKMGLQNYLQAQ WRCRNCGYVHEGTGAPELCPACAHPKAHFELLGINW. :. I REE
45 Sequence Alignment Correction PHE ASP ILE CYS ARG LEU PRO GLY SER ALA GLU ALA VAL CYS TEMPLATE PHE ASN VAL CYS ARG THR PRO GLU ALA ILE CYS TARGET (ALIGNMENT 1) PHE ASN VAL CYS ARG THR PRO GLU ALA ILE CYS TARGET (ALIGNMENT 2) "Alignment 1" is chosen because of the PROs at position 7. But the 10 Angstrom gap that results is too big to close with a single peptide bond.
46 A Fold Recognition Example - 3D-PSSM 3D-PSSM combines: Target sequence profiles. Template sequence profiles. Residue equivalence. Secondary structure matching. Solvation potentials. Sequences are aligned to folds using dynamic programming with the alignments scored by a range of 1D and 3D profiles.
47 3D-PSSM Fold Profile Library PSI-BLAST and a non-redundant database are used to create profiles for each of the folds in the library. Each fold is aligned with members of the same superfamily using the structural alignment program SSAP. Those folds from SCOP with sufficient structural similarity are then also used to create profiles using PSI-BLAST in the same way. All the related profiles are merged using the structural alignment to form a 3D-profile.
48 Secondary Structure and Solvation Potentials Secondary structure is assigned to each fold based on the annotation in the STRIDE database. Each residue in the fold is also assigned a solvation potential. The degree of burial of each residue is defined as the ratio between its solvent accessible surface area and its overall surface area. Solvation potential is divided into 21 bins, ranging from 0% (buried) to 100%(exposed).
49 Sequence and Secondary Structure Profiles 3D-PSSM also uses the coincidence of predicted secondary structure (target sequence) and known secondary structure (fold). Here a simple scoring scheme is used for matching secondary structure types, +1 for a match, otherwise -1.
50 Preparing the Query Sequence Query sequences have their secondary structure predicted by PSI-Pred. PSI-BLAST profiles are also generated for the query sequence to allow bi-directional scoring. The 3D-FSSP dynamic programming algorithm is used to scan the fold library with the query sequence.
51 3D-PSSM - Dynamic Programming Three passes of dynamic programming are performed for each querytemplate alignment. Each pass uses a different matrix to score the alignment, but secondary structure and solvation potential are used in each pass. The score for a match between a query residue and a fold residue is calculated the sum of the secondary structure, solvation potential and profile scores. The final score is simply the maximum of the scores from the three passes.
52 Differences between profile-based methods (Rychlewski( Rychlewski,, et al, 2000) PSI-BLAST PDB-BLAST Multiple alignments: 5 iterations with 10-3 evalue treshold Profile: Preclustering with 98% cutoff, pseudocount based on variability estimation-background aminoacid frequencies Database: NR Multiple alignment: same as PSI-Blast Profile: same as PSI-Blast Database: PDB database BASIC Multiple alignment: 2 PSI-Blast it. with 0.1 e-value threshold Profile: preclustering with 97% id cutoff; amino-acid composition filter, distant homologues have smaller weights Database: profiles of proteins from PDB FFAS/FFAS03 Multiple alignment: same as PSI-Blast Profile: preclustering with 97% id cutoff; amino-acid composition filter, sequence diversity based weight Database: profiles of proteins from PDB

53 Baker & Sali, Science 2001.
54 COMBINING ADDITIONAL INFORMATION Conserved Tree-Determinant Correlated mutations




55 rcc1 ran Ras Ral Rho Ras Ral Rho by J.A. G-Ranea

56 Azuma et al., J,Mol. Biol. 1999
 Model GDP E157 H304 Mg++")

57 Complex (Model on Vomplex superposition) Mapping of mutants (side view) Model GDP E157 H304 Mg++ D44 H410 H78 E157 H270 Mg++ GDP D44 H304 R206 H78 R206 H410 H270 D128 D128 H78 Green: Km, red: Kcat.


58 VISUALIZATION Pazos et al.,
59 Fold Recognition Servers I 3D-PSSM - Based on sequence profiles, solvatation potentials and secondary structure. SPARKS2 - Top server in CM predictions in CASP 6. Sequence, secondary structure Profiles And Residue-level Knowledgebased Score for fold recognition. mgenthreader - Combines profiles and sequence-structure alignments. A neural network-based jury system calculates the final score based on solvation and pair potentials.
60 Fold Recognition Servers I RAPTOR - Best-scoring server in CAFASP3 competition in You have to ask to use it first... ROBETTA - ROBETTA makes both ab initio and template-based predictions. It detects fragments with BLAST, FFAS03, or 3DJury, generates alignments with its own K*SYNC method and uses fragment insertion and assembly. PHYRE - A new server (so new it doesnt even have documentation that attempts to assemble fragments in a similar way to Robetta.
61 Advanced Sequence-Based and Hybrid Techniques PSIBLAST Profile methods, beginning with PSI-Blast, can be as accurate as many fold recognition techniques at detecting remote homologues. Although expert users of these methods can usually spot biologically meaningful templates from careful analysis of low-scoring hits, many remote homologues are not detected. Intermediate Searching Profile-profile alignment methods use evolutionary information in both query and template sequences. As a result, they are able to detect remote homologies beyond the reach of other sequence comparison methods. Hhpred! Profile-profile
62 Advanced Sequence-Based and Hybrid Techniques Hidden Markov Models Hidden Markov models were originally developed for speech recognition. They regard the sequence as a series of nodes, each corresponding to a column in a multiple alignment. Each node has a residue state and states for insertion and deletion. A model can be built from many sequences and these models have many similarities to profiles. META-PROFILES Many methods now also use predicted secondary structure. By adding structural information to the profiles (metaprofiles) it is often possible to find homologues that have very low sequence similarity but are still structurally similar..
63 Hybrid Sequence-Based Servers SAM T The query is checked against a library of hidden Markov models. This is NOT a threading technique, it is sequence based, but it does use secondary structure information. Meta-BASIC - basic.bioinfo.pl Meta-BASIC is based on consensus alignments of profiles. It combines sequence profiles with predicted secondary structure and uses several scoring systems and alignment algorithms. FFAS ffas.ljcrf.edu/ffas-cgi/cgi/ffas.pl FFAS03 is a profile-profile alignment method, which takes advantage of the evolutionary information in both query and template sequences.
64 Consensus Fold Recognition It has long been recognised that human experts are better at fold prediction than the methods these same experts had developed. Human experts usually use several different fold recognition methods and predict folds after evaluating all the results (not just the top hits) from a range of methods. So why not produce an algorithm that mimics the human experts? In first consensus server, Pcons, the target sequence was sent to six publicly available fold recognition web servers. Models were built from all the predictions. The models were then structurally superimposed and evaluated for their similarity. The quality of the model was predicted from the rescaled score and from its similarity to other predicted models.
65 Consensus Fold Recognition Servers 3D Jury - 3D Jury is a consensus predictor that utilizes the results of fold recognition servers, such as FFAS, 3D-PSSM, FUGUE and mgenthreader, and uses a jury system to select structures INGBU - This produces a consensus prediction based on five methods that exploit sequence and structure information in different ways. Pcons - Pcons was the first consensus server for fold recognition. It selects the best prediction from several servers. PMOD can also generate models using the alignment, template and MODELLER
66 Structure Prediction in a Nutshell Target sequence Biological information from papers Active sites, domains, cofactors etc. Are there domains? PFAM/ProDom/ InterPro - BLAST results Secondary structure, accessibility, Trans-membrane segments PHD, PSIPRED Domain1 Domain 2 Domain 3 etc... BLAST search for PDB Structural Template Yes No Homology modelling programs SWISSMODEL, coremodeller Align with template Consenus Servers, 3D Jury Alignment 1 Alignment 2 Alignment 3 Alignment 4... Loops... Fold Recognition Servers Eg 3DPSSM GenTHREADER Model Evaluation 3D - ProSa model Ana Rojas - Biotech Mendoza Structural Bioinformatics suite Group Side chain canonical Complete loops MaxSprout 3D model
67 SOME REAL EXAMPLES BIOLOGICALLY RELEVANT
68 PAAD DOMAIN AIM: TRY TO PREDICT BINDING MODE structure was unknown: we needed a model.
69 BACKGROUND WHERE IS THE PAAD DOMAIN? 1.-First, location of this domain using BLAST! PAAD family: MEFV/PYRIN (Pawlowski, et.al., 2001, others) Nacht family: PAN/NALPs/DEFCAP/PYCARD, CATERPILLER (Tschopp et al, Nature, 2003)


70 BACKGROUND THE PROBLEM OF DOMAIN SHUFFLING NALP2 PAAD NACHT LRR S ASC2 PAAD MATER? NACHT LRR S ASC PAAD CARD CARD4 CARD NACHT LRR S CASPASE ZF PAAD CASPASE NOD2 CARD CARD NACHT LRR S PYRIN PAAD B-BOX Zn FINGER SPRY NAIP BIR BIR BIR NACHT LRR S COS1.5? NACHT LRR S IF16 PAAD IF120X IF120X CLAN CARD NACHT LRR S MNDA,AIM2 PAAD IF120X NAC PAAD? NACHT LRR S? CARD Sensors! They connect different pathways! 2.-Domain analyses in different sequences (PFAM)
 PAAD")
71 WHERE DOES IT COME FROM? 3.-Phylogenetic analyses (PFAM) PAAD CARD DD DED

72 Hydrophobic core (sol. acc. area <10% maximum solv. area) 4.-MAL & Sec. Structure Prediction HELIX 3 does not have core residues. In DD, and others helix3 doesn t pack too well

73 domain Homology modeling of PAAD domain (MEFV from mouse) N N H3 H3 C C 4.-Template detection, alignment and modeling! Hydrophobic core



74 pyrin LYS35 LYS52 LYS39 ARG49 ARG ILE40 PRO41 VAL51 MET45 Charged patch Pan2/NALP4 Hydrophobic patch 4.-Identification of patches or relevant features in the surfaces! ALA50 TRP44 LYS48 VAL47 PRO43 ILE42



 + CHARGED")
75 IFI204 ASP32 LYS64 90 o GLU53 GLU71 GLU67 GLU70 GLU54 LYS76 LYS55 AIM2 ASP19 LYS23 GLU o ARG67 LYS71 LYS64 - CHARGED (CONCAVE) + CHARGED (CONVEX) +CHARGED

76 Paad is a 6 alpha helical bundle Helix 3 is disordered Binding patches correctly predicted Real structure 1PN5 Released October 2003 September 2003

77 SPOC DOMAIN Combining HMMER sequence analyses and threading
78 METHODS: Selecting regions first! Query seq Blast to nr/uniprot90 Blast to EST s & unfinished genomes Multiple alignment T COFFEE, MUSCLE, etc TO ENRICH PROFILE! PROFILE BUILDING HMMER/PSI BLAST SEARCHES in Uniprot90
79 METHODS: HMMER Strategy/Intermediate searches Known Known!!!
80 METHODS HMMER ANALYSES III iso1 iso aa NLS PHD 614 aa Coiled coil SPOC: Protein protein interaction (Sanchez Pulido et al, 2004) iso aa




81 METHODS HMMER ANALYSES III iso2 SPOC: Protein protein interaction RBMF_HUMAN Homology Structural modeling Bioinformatics Group
82 Acknowledgments Michael Tress, David de Juan (CNIO) Florencio Pazos, Luis Sanchez-Pulido (CNB) Rest of (CNIO) and anyone else whose figures I used...
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Programme Last week s quiz results + Summary Fold recognition Break Exercise: Modelling remote homologues
 Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Protein Structure Prediction
 Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Sequence Analysis and Databases 2: Sequences and Multiple Alignments
 1 Sequence Analysis and Databases 2: Sequences and Multiple Alignments Jose María González-Izarzugaza Martínez CNIO Spanish National Cancer Research Centre (jmgonzalez@cnio.es) 2 Sequence Comparisons:
1 Sequence Analysis and Databases 2: Sequences and Multiple Alignments Jose María González-Izarzugaza Martínez CNIO Spanish National Cancer Research Centre (jmgonzalez@cnio.es) 2 Sequence Comparisons:
Packing of Secondary Structures
 7.88 Lecture Notes - 4 7.24/7.88J/5.48J The Protein Folding and Human Disease Professor Gossard Retrieving, Viewing Protein Structures from the Protein Data Base Helix helix packing Packing of Secondary
7.88 Lecture Notes - 4 7.24/7.88J/5.48J The Protein Folding and Human Disease Professor Gossard Retrieving, Viewing Protein Structures from the Protein Data Base Helix helix packing Packing of Secondary
Bioinformatics: Secondary Structure Prediction
 Bioinformatics: Secondary Structure Prediction Prof. David Jones d.jones@cs.ucl.ac.uk LMLSTQNPALLKRNIIYWNNVALLWEAGSD The greatest unsolved problem in molecular biology:the Protein Folding Problem? Entries
Bioinformatics: Secondary Structure Prediction Prof. David Jones d.jones@cs.ucl.ac.uk LMLSTQNPALLKRNIIYWNNVALLWEAGSD The greatest unsolved problem in molecular biology:the Protein Folding Problem? Entries
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Sequence Alignments. Dynamic programming approaches, scoring, and significance. Lucy Skrabanek ICB, WMC January 31, 2013
 Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer Protein Structure Prediction I
 BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 9. Protein Structure Prediction I Structure Prediction Overview Overview of problem variants Secondary structure prediction
BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 9. Protein Structure Prediction I Structure Prediction Overview Overview of problem variants Secondary structure prediction
Identification of correct regions in protein models using structural, alignment, and consensus information
 Identification of correct regions in protein models using structural, alignment, and consensus information BJO RN WALLNER AND ARNE ELOFSSON Stockholm Bioinformatics Center, Stockholm University, SE-106
Identification of correct regions in protein models using structural, alignment, and consensus information BJO RN WALLNER AND ARNE ELOFSSON Stockholm Bioinformatics Center, Stockholm University, SE-106
Protein structure alignments
 Protein structure alignments Proteins that fold in the same way, i.e. have the same fold are often homologs. Structure evolves slower than sequence Sequence is less conserved than structure If BLAST gives
Protein structure alignments Proteins that fold in the same way, i.e. have the same fold are often homologs. Structure evolves slower than sequence Sequence is less conserved than structure If BLAST gives
1-D Predictions. Prediction of local features: Secondary structure & surface exposure
 1-D Predictions Prediction of local features: Secondary structure & surface exposure 1 Learning Objectives After today s session you should be able to: Explain the meaning and usage of the following local
1-D Predictions Prediction of local features: Secondary structure & surface exposure 1 Learning Objectives After today s session you should be able to: Explain the meaning and usage of the following local
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
Steps in protein modelling. Structure prediction, fold recognition and homology modelling. Basic principles of protein structure
 Structure prediction, fold recognition and homology modelling Marjolein Thunnissen Lund September 2012 Steps in protein modelling 3-D structure known Comparative Modelling Sequence of interest Similarity
Structure prediction, fold recognition and homology modelling Marjolein Thunnissen Lund September 2012 Steps in protein modelling 3-D structure known Comparative Modelling Sequence of interest Similarity
SCOP. all-β class. all-α class, 3 different folds. T4 endonuclease V. 4-helical cytokines. Globin-like
 SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
Neural Networks for Protein Structure Prediction Brown, JMB CS 466 Saurabh Sinha
 Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
Motifs, Profiles and Domains. Michael Tress Protein Design Group Centro Nacional de Biotecnología, CSIC
 Motifs, Profiles and Domains Michael Tress Protein Design Group Centro Nacional de Biotecnología, CSIC Comparing Two Proteins Sequence Alignment Determining the pattern of evolution and identifying conserved
Motifs, Profiles and Domains Michael Tress Protein Design Group Centro Nacional de Biotecnología, CSIC Comparing Two Proteins Sequence Alignment Determining the pattern of evolution and identifying conserved
Building 3D models of proteins
 Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
09/06/25. Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Non-uniform distribution of folds. Scheme of protein structure predicition
 Non-uniform distribution of folds. Scheme of protein structure predicition") Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
Bioinformatics: Secondary Structure Prediction
 Bioinformatics: Secondary Structure Prediction Prof. David Jones d.t.jones@ucl.ac.uk Possibly the greatest unsolved problem in molecular biology: The Protein Folding Problem MWMPPRPEEVARK LRRLGFVERMAKG
Bioinformatics: Secondary Structure Prediction Prof. David Jones d.t.jones@ucl.ac.uk Possibly the greatest unsolved problem in molecular biology: The Protein Folding Problem MWMPPRPEEVARK LRRLGFVERMAKG
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics
 Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program)
") Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Structure and evolution of the spliceosomal peptidyl-prolyl cistrans isomerase Cwc27
 Acta Cryst. (2014). D70, doi:10.1107/s1399004714021695 Supporting information Volume 70 (2014) Supporting information for article: Structure and evolution of the spliceosomal peptidyl-prolyl cistrans isomerase
Acta Cryst. (2014). D70, doi:10.1107/s1399004714021695 Supporting information Volume 70 (2014) Supporting information for article: Structure and evolution of the spliceosomal peptidyl-prolyl cistrans isomerase
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Protein Structures: Experiments and Modeling. Patrice Koehl
 Protein Structures: Experiments and Modeling Patrice Koehl Structural Bioinformatics: Proteins Proteins: Sources of Structure Information Proteins: Homology Modeling Proteins: Ab initio prediction Proteins:
Protein Structures: Experiments and Modeling Patrice Koehl Structural Bioinformatics: Proteins Proteins: Sources of Structure Information Proteins: Homology Modeling Proteins: Ab initio prediction Proteins:
Physiochemical Properties of Residues
 Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009
 114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Protein Bioinformatics. Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet sandberg.cmb.ki.
 Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
3D Structure. Prediction & Assessment Pt. 2. David Wishart 3-41 Athabasca Hall
 3D Structure Prediction & Assessment Pt. 2 David Wishart 3-41 Athabasca Hall david.wishart@ualberta.ca Objectives Become familiar with methods and algorithms for secondary Structure Prediction Become familiar
3D Structure Prediction & Assessment Pt. 2 David Wishart 3-41 Athabasca Hall david.wishart@ualberta.ca Objectives Become familiar with methods and algorithms for secondary Structure Prediction Become familiar
HMM applications. Applications of HMMs. Gene finding with HMMs. Using the gene finder
 HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
CS612 - Algorithms in Bioinformatics
 Fall 2017 Protein Structure Detection Methods October 30, 2017 Comparative Modeling Comparative modeling is modeling of the unknown based on comparison to what is known In the context of modeling or computing
Fall 2017 Protein Structure Detection Methods October 30, 2017 Comparative Modeling Comparative modeling is modeling of the unknown based on comparison to what is known In the context of modeling or computing
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5
 Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
PROTEIN SECONDARY STRUCTURE PREDICTION: AN APPLICATION OF CHOU-FASMAN ALGORITHM IN A HYPOTHETICAL PROTEIN OF SARS VIRUS
 Int. J. LifeSc. Bt & Pharm. Res. 2012 Kaladhar, 2012 Research Paper ISSN 2250-3137 www.ijlbpr.com Vol.1, Issue. 1, January 2012 2012 IJLBPR. All Rights Reserved PROTEIN SECONDARY STRUCTURE PREDICTION:
Int. J. LifeSc. Bt & Pharm. Res. 2012 Kaladhar, 2012 Research Paper ISSN 2250-3137 www.ijlbpr.com Vol.1, Issue. 1, January 2012 2012 IJLBPR. All Rights Reserved PROTEIN SECONDARY STRUCTURE PREDICTION:
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Protein Secondary Structure Prediction using Feed-Forward Neural Network
 COPYRIGHT 2010 JCIT, ISSN 2078-5828 (PRINT), ISSN 2218-5224 (ONLINE), VOLUME 01, ISSUE 01, MANUSCRIPT CODE: 100713 Protein Secondary Structure Prediction using Feed-Forward Neural Network M. A. Mottalib,
COPYRIGHT 2010 JCIT, ISSN 2078-5828 (PRINT), ISSN 2218-5224 (ONLINE), VOLUME 01, ISSUE 01, MANUSCRIPT CODE: 100713 Protein Secondary Structure Prediction using Feed-Forward Neural Network M. A. Mottalib,
Supplementary Figure 3 a. Structural comparison between the two determined structures for the IL 23:MA12 complex. The overall RMSD between the two
 Supplementary Figure 1. Biopanningg and clone enrichment of Alphabody binders against human IL 23. Positive clones in i phage ELISA with optical density (OD) 3 times higher than background are shown for
Supplementary Figure 1. Biopanningg and clone enrichment of Alphabody binders against human IL 23. Positive clones in i phage ELISA with optical density (OD) 3 times higher than background are shown for
Secondary Structure. Bioch/BIMS 503 Lecture 2. Structure and Function of Proteins. Further Reading. Φ, Ψ angles alone determine protein structure
 Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Protein Structure Prediction using String Kernels. Technical Report
 Protein Structure Prediction using String Kernels Technical Report Department of Computer Science and Engineering University of Minnesota 4-192 EECS Building 200 Union Street SE Minneapolis, MN 55455-0159
Protein Structure Prediction using String Kernels Technical Report Department of Computer Science and Engineering University of Minnesota 4-192 EECS Building 200 Union Street SE Minneapolis, MN 55455-0159
Christian Sigrist. November 14 Protein Bioinformatics: Sequence-Structure-Function 2018 Basel
 Christian Sigrist General Definition on Conserved Regions Conserved regions in proteins can be classified into 5 different groups: Domains: specific combination of secondary structures organized into a
Christian Sigrist General Definition on Conserved Regions Conserved regions in proteins can be classified into 5 different groups: Domains: specific combination of secondary structures organized into a
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Protein Structure Prediction, Engineering & Design CHEM 430
 Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
SUPPLEMENTARY MATERIALS
 SUPPLEMENTARY MATERIALS Enhanced Recognition of Transmembrane Protein Domains with Prediction-based Structural Profiles Baoqiang Cao, Aleksey Porollo, Rafal Adamczak, Mark Jarrell and Jaroslaw Meller Contact:
SUPPLEMENTARY MATERIALS Enhanced Recognition of Transmembrane Protein Domains with Prediction-based Structural Profiles Baoqiang Cao, Aleksey Porollo, Rafal Adamczak, Mark Jarrell and Jaroslaw Meller Contact:
Structural Alignment of Proteins
 Goal Align protein structures Structural Alignment of Proteins 1 2 3 4 5 6 7 8 9 10 11 12 13 14 PHE ASP ILE CYS ARG LEU PRO GLY SER ALA GLU ALA VAL CYS PHE ASN VAL CYS ARG THR PRO --- --- --- GLU ALA ILE
Goal Align protein structures Structural Alignment of Proteins 1 2 3 4 5 6 7 8 9 10 11 12 13 14 PHE ASP ILE CYS ARG LEU PRO GLY SER ALA GLU ALA VAL CYS PHE ASN VAL CYS ARG THR PRO --- --- --- GLU ALA ILE
Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction. Sepp Hochreiter
 Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction Institute of Bioinformatics Johannes Kepler University, Linz, Austria Chapter 4 Protein Secondary
Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction Institute of Bioinformatics Johannes Kepler University, Linz, Austria Chapter 4 Protein Secondary
Protein structure. Protein structure. Amino acid residue. Cell communication channel. Bioinformatics Methods
 Cell communication channel Bioinformatics Methods Iosif Vaisman Email: ivaisman@gmu.edu SEQUENCE STRUCTURE DNA Sequence Protein Sequence Protein Structure Protein structure ATGAAATTTGGAAACTTCCTTCTCACTTATCAGCCACCT...
Cell communication channel Bioinformatics Methods Iosif Vaisman Email: ivaisman@gmu.edu SEQUENCE STRUCTURE DNA Sequence Protein Sequence Protein Structure Protein structure ATGAAATTTGGAAACTTCCTTCTCACTTATCAGCCACCT...
Protein Structure Prediction and Display
 Protein Structure Prediction and Display Goal Take primary structure (sequence) and, using rules derived from known structures, predict the secondary structure that is most likely to be adopted by each
Protein Structure Prediction and Display Goal Take primary structure (sequence) and, using rules derived from known structures, predict the secondary structure that is most likely to be adopted by each
Can protein model accuracy be. identified? NO! CBS, BioCentrum, Morten Nielsen, DTU
 Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Computational Molecular Biology. Protein Structure and Homology Modeling
 Computational Molecular Biology Protein Structure and Homology Modeling Prof. Alejandro Giorge1 Dr. Francesco Musiani Sequence, function and structure relationships v Life is the ability to metabolize
Computational Molecular Biology Protein Structure and Homology Modeling Prof. Alejandro Giorge1 Dr. Francesco Musiani Sequence, function and structure relationships v Life is the ability to metabolize
Bioinformatics Practical for Biochemists
 Bioinformatics Practical for Biochemists Andrei Lupas, Birte Höcker, Steffen Schmidt WS 2013/14 03. Sequence Features Targeting proteins signal peptide targets proteins to the secretory pathway N-terminal
Bioinformatics Practical for Biochemists Andrei Lupas, Birte Höcker, Steffen Schmidt WS 2013/14 03. Sequence Features Targeting proteins signal peptide targets proteins to the secretory pathway N-terminal
We used the PSI-BLAST program (http://www.ncbi.nlm.nih.gov/blast/) to search the
 to search the") SUPPLEMENTARY METHODS - in silico protein analysis We used the PSI-BLAST program (http://www.ncbi.nlm.nih.gov/blast/) to search the Protein Data Bank (PDB, http://www.rcsb.org/pdb/) and the NCBI non-redundant
SUPPLEMENTARY METHODS - in silico protein analysis We used the PSI-BLAST program (http://www.ncbi.nlm.nih.gov/blast/) to search the Protein Data Bank (PDB, http://www.rcsb.org/pdb/) and the NCBI non-redundant
Similarity or Identity? When are molecules similar?
 Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Similarity searching summary (2)
") Similarity searching / sequence alignment summary Biol4230 Thurs, February 22, 2016 Bill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 What have we covered? Homology excess similiarity but no excess similarity
Similarity searching / sequence alignment summary Biol4230 Thurs, February 22, 2016 Bill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 What have we covered? Homology excess similiarity but no excess similarity
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
Bioinformatics. Proteins II. - Pattern, Profile, & Structure Database Searching. Robert Latek, Ph.D. Bioinformatics, Biocomputing
 Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Template-Based Modeling of Protein Structure
 Template-Based Modeling of Protein Structure David Constant Biochemistry 218 December 11, 2011 Introduction. Much can be learned about the biology of a protein from its structure. Simply put, structure
Template-Based Modeling of Protein Structure David Constant Biochemistry 218 December 11, 2011 Introduction. Much can be learned about the biology of a protein from its structure. Simply put, structure
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
COMP 598 Advanced Computational Biology Methods & Research. Introduction. Jérôme Waldispühl School of Computer Science McGill University
 COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
proteins Refinement by shifting secondary structure elements improves sequence alignments
 proteins STRUCTURE O FUNCTION O BIOINFORMATICS Refinement by shifting secondary structure elements improves sequence alignments Jing Tong, 1,2 Jimin Pei, 3 Zbyszek Otwinowski, 1,2 and Nick V. Grishin 1,2,3
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Refinement by shifting secondary structure elements improves sequence alignments Jing Tong, 1,2 Jimin Pei, 3 Zbyszek Otwinowski, 1,2 and Nick V. Grishin 1,2,3
Introduction to Evolutionary Concepts
 Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2009 VMD/MultiSeq
Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2009 VMD/MultiSeq
Procheck output. Bond angles (Procheck) Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.
 Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.") Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Basics of protein structure
 Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Supplemental Materials for. Structural Diversity of Protein Segments Follows a Power-law Distribution
 Supplemental Materials for Structural Diversity of Protein Segments Follows a Power-law Distribution Yoshito SAWADA and Shinya HONDA* National Institute of Advanced Industrial Science and Technology (AIST),
Supplemental Materials for Structural Diversity of Protein Segments Follows a Power-law Distribution Yoshito SAWADA and Shinya HONDA* National Institute of Advanced Industrial Science and Technology (AIST),
Genomics and bioinformatics summary. Finding genes -- computer searches
 Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Protein Secondary Structure Assignment and Prediction
 1 Protein Secondary Structure Assignment and Prediction Defining SS features - Dihedral angles, alpha helix, beta stand (Hydrogen bonds) Assigned manually by crystallographers or Automatic DSSP (Kabsch
1 Protein Secondary Structure Assignment and Prediction Defining SS features - Dihedral angles, alpha helix, beta stand (Hydrogen bonds) Assigned manually by crystallographers or Automatic DSSP (Kabsch
Intro Secondary structure Transmembrane proteins Function End. Last time. Domains Hidden Markov Models
 Last time Domains Hidden Markov Models Today Secondary structure Transmembrane proteins Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL
Last time Domains Hidden Markov Models Today Secondary structure Transmembrane proteins Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL
Structure to Function. Molecular Bioinformatics, X3, 2006
 Structure to Function Molecular Bioinformatics, X3, 2006 Structural GeNOMICS Structural Genomics project aims at determination of 3D structures of all proteins: - organize known proteins into families
Structure to Function Molecular Bioinformatics, X3, 2006 Structural GeNOMICS Structural Genomics project aims at determination of 3D structures of all proteins: - organize known proteins into families
Protein Structure Prediction
 Protein Structure Prediction Michael Feig MMTSB/CTBP 2006 Summer Workshop From Sequence to Structure SEALGDTIVKNA Ab initio Structure Prediction Protocol Amino Acid Sequence Conformational Sampling to
Protein Structure Prediction Michael Feig MMTSB/CTBP 2006 Summer Workshop From Sequence to Structure SEALGDTIVKNA Ab initio Structure Prediction Protocol Amino Acid Sequence Conformational Sampling to
Today. Last time. Secondary structure Transmembrane proteins. Domains Hidden Markov Models. Structure prediction. Secondary structure
 Last time Today Domains Hidden Markov Models Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL SSLGPVVDAHPEYEEVALLERMVIPERVIE FRVPWEDDNGKVHVNTGYRVQFNGAIGPYK
Last time Today Domains Hidden Markov Models Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL SSLGPVVDAHPEYEEVALLERMVIPERVIE FRVPWEDDNGKVHVNTGYRVQFNGAIGPYK
Properties of amino acids in proteins
 Properties of amino acids in proteins one of the primary roles of DNA (but not the only one!) is to code for proteins A typical bacterium builds thousands types of proteins, all from ~20 amino acids repeated
Properties of amino acids in proteins one of the primary roles of DNA (but not the only one!) is to code for proteins A typical bacterium builds thousands types of proteins, all from ~20 amino acids repeated
IT og Sundhed 2010/11
 IT og Sundhed 2010/11 Sequence based predictors. Secondary structure and surface accessibility Bent Petersen 13 January 2011 1 NetSurfP Real Value Solvent Accessibility predictions with amino acid associated
IT og Sundhed 2010/11 Sequence based predictors. Secondary structure and surface accessibility Bent Petersen 13 January 2011 1 NetSurfP Real Value Solvent Accessibility predictions with amino acid associated
Supersecondary Structures (structural motifs)
") Supersecondary Structures (structural motifs) Various Sources Slide 1 Supersecondary Structures (Motifs) Supersecondary Structures (Motifs): : Combinations of secondary structures in specific geometric
Supersecondary Structures (structural motifs) Various Sources Slide 1 Supersecondary Structures (Motifs) Supersecondary Structures (Motifs): : Combinations of secondary structures in specific geometric
2MHR. Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity.
 Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
CSE 549: Computational Biology. Substitution Matrices
 CSE 9: Computational Biology Substitution Matrices How should we score alignments So far, we ve looked at arbitrary schemes for scoring mutations. How can we assign scores in a more meaningful way? Are
CSE 9: Computational Biology Substitution Matrices How should we score alignments So far, we ve looked at arbitrary schemes for scoring mutations. How can we assign scores in a more meaningful way? Are
Major Types of Association of Proteins with Cell Membranes. From Alberts et al
 Major Types of Association of Proteins with Cell Membranes From Alberts et al Proteins Are Polymers of Amino Acids Peptide Bond Formation Amino Acid central carbon atom to which are attached amino group
Major Types of Association of Proteins with Cell Membranes From Alberts et al Proteins Are Polymers of Amino Acids Peptide Bond Formation Amino Acid central carbon atom to which are attached amino group
PROTEIN STRUCTURE PREDICTION Bioinformatic Approach
 Link to Order: http://fivephoton.com/index.php?route=product/product&path=37&product_id=55 Price: $109.95 Website:. PROTEIN STRUCTURE PREDICTION Bioinformatic Approach edited by IGOR F. TSIGELNY Table
Link to Order: http://fivephoton.com/index.php?route=product/product&path=37&product_id=55 Price: $109.95 Website:. PROTEIN STRUCTURE PREDICTION Bioinformatic Approach edited by IGOR F. TSIGELNY Table
Modeling for 3D structure prediction
 Modeling for 3D structure prediction What is a predicted structure? A structure that is constructed using as the sole source of information data obtained from computer based data-mining. However, mixing
Modeling for 3D structure prediction What is a predicted structure? A structure that is constructed using as the sole source of information data obtained from computer based data-mining. However, mixing
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Bioinformatics Algorithms Hidden Markov Models
 Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Sequence Based Bioinformatics
 Structural and Functional Analysis of Inosine Monophosphate Dehydrogenase using Sequence-Based Bioinformatics Barry Sexton 1,2 and Troy Wymore 3 1 Bioengineering and Bioinformatics Summer Institute, Department
Structural and Functional Analysis of Inosine Monophosphate Dehydrogenase using Sequence-Based Bioinformatics Barry Sexton 1,2 and Troy Wymore 3 1 Bioengineering and Bioinformatics Summer Institute, Department
Protein Structure Determination
 Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Conditional Graphical Models
 PhD Thesis Proposal Conditional Graphical Models for Protein Structure Prediction Yan Liu Language Technologies Institute University Thesis Committee Jaime Carbonell (Chair) John Lafferty Eric P. Xing
PhD Thesis Proposal Conditional Graphical Models for Protein Structure Prediction Yan Liu Language Technologies Institute University Thesis Committee Jaime Carbonell (Chair) John Lafferty Eric P. Xing
Course Notes: Topics in Computational. Structural Biology.
 Course Notes: Topics in Computational Structural Biology. Bruce R. Donald June, 2010 Copyright c 2012 Contents 11 Computational Protein Design 1 11.1 Introduction.........................................
Course Notes: Topics in Computational Structural Biology. Bruce R. Donald June, 2010 Copyright c 2012 Contents 11 Computational Protein Design 1 11.1 Introduction.........................................
Molecular Modeling Lecture 7. Homology modeling insertions/deletions manual realignment
 Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
Heteropolymer. Mostly in regular secondary structure
 Heteropolymer - + + - Mostly in regular secondary structure 1 2 3 4 C >N trace how you go around the helix C >N C2 >N6 C1 >N5 What s the pattern? Ci>Ni+? 5 6 move around not quite 120 "#$%&'!()*(+2!3/'!4#5'!1/,#64!#6!,6!
Heteropolymer - + + - Mostly in regular secondary structure 1 2 3 4 C >N trace how you go around the helix C >N C2 >N6 C1 >N5 What s the pattern? Ci>Ni+? 5 6 move around not quite 120 "#$%&'!()*(+2!3/'!4#5'!1/,#64!#6!,6!
Peptides And Proteins
 Kevin Burgess, May 3, 2017 1 Peptides And Proteins from chapter(s) in the recommended text A. Introduction B. omenclature And Conventions by amide bonds. on the left, right. 2 -terminal C-terminal triglycine
Kevin Burgess, May 3, 2017 1 Peptides And Proteins from chapter(s) in the recommended text A. Introduction B. omenclature And Conventions by amide bonds. on the left, right. 2 -terminal C-terminal triglycine
Getting To Know Your Protein
 Getting To Know Your Protein Comparative Protein Analysis: Part III. Protein Structure Prediction and Comparison Robert Latek, PhD Sr. Bioinformatics Scientist Whitehead Institute for Biomedical Research
Getting To Know Your Protein Comparative Protein Analysis: Part III. Protein Structure Prediction and Comparison Robert Latek, PhD Sr. Bioinformatics Scientist Whitehead Institute for Biomedical Research
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of