PGA: A Program for Genome Annotation by Comparative Analysis of. Maximum Likelihood Phylogenies of Genes and Species
|
|
|
- Frank Dalton
- 6 years ago
- Views:
Transcription
1 PGA: A Program for Genome Annotation by Comparative Analysis of Maximum Likelihood Phylogenies of Genes and Species Paulo Bandiera-Paiva 1 and Marcelo R.S. Briones 2 1 Departmento de Informática em Saúde and 2 Departamento de Microbiologia, Imunologia e Parasitologia, Universidade Federal de São Paulo, , São Paulo, Brazil. To whom correspondence should be addressed Paulo Bandiera-Paiva Ed. Leal Prado, Universidade Federal de São Paulo, Rua Botucatu, 862, Térreo. CEP , São Paulo, S.P., Brazil paiva@unifesp.br Tel: (55)(11) FAX: (55)(11)
2 ABSTRACT The Phylogenetic Genome Annotator (PGA) is a computer program that enables real-time comparison of gene trees versus species trees obtained from predicted open reading frames of whole genome data. The gene phylogenies are inferred for each individual genome predicted proteins whereas the species phylogenies are inferred from rdna data. The correlated protein domains, defined by PFAM, are then displayed side-by-side with a phylogeny of the corresponding species. The statistical support of gene clusters (branches) is given by the quartet puzzling method. This analysis readily discriminates paralogs from orthologs, enabling the identification of proteins originated by gene duplications and the prediction of possible functional divergence in groups of similar sequences. Availability: PGA and documentation is freely available under the GNU General Public License and can be downloaded from: Contact: pga@compbio.epm.br 2
3 INTRODUCTION The characterization of distinct protein functions in a group of sequences sharing a common domain can be improved by comparing the evolutionary history of the underlying species with the inferred phylogeny of the genes (Eisen, 1998). Differences between topologies of these two phylogenies indicate gene duplication events. The prediction of protein function from primary structure is a common task that can be performed using different methodologies. The most common approach aims to identify similarities between the query sequence and sequences with known function, assuming that similar sequences share a common ancestor and thus include stretches of similar states also present in the precursor sequence. This assumption of orthology based on similarity, disregards the possibility of gene duplication (paralogy) or convergent evolution of the genes. The comparison between gene trees versus species trees is informative about the evolutionary history of genes under study, resulting in a more reliable annotation of predicted proteins of unknown function. An integrated tool for the analysis of correlated proteins that share a common domain is presented here. This program, PGA, explicitly considers the phylogenies of species and predicted proteins which enables the visualization of gene duplication, or paralogy, and therefore contributing to reveal novel functions in a group of similar sequences. DESCRIPTION The phylogenetic genome annotator (PGA) uses two coordinated datasets, one of proteins (gene sequence set) and the other of ribosomal RNA from their underlying species (species sequence set). The first dataset is used for inferring the phylogeny of the genes, whereas the second for the inference of the species tree of the source organisms bearing the proteins of the first dataset. The gene sequence sets are data structures representing groups of related sequences, 3
4 containing taxonomic information, primary structure, and a multiple alignment. This structure is used both for protein sequences and to access nucleotide sequences of ribosomal RNA, used for the inference of species phylogeny. The gene sequence set is used for the manipulation of protein sequences representing genes of interest, while the species sequence set maintains the rrna sequences from the source organisms present in the prior set. The gene sequence set is specified by the user from a group of related proteins. These sequences may be manually entered, specified by accession id, or automatically fetched from the seed sequences of a specified PFAM protein domain family (Bateman et al., 2000). Proteins specified by accession and those pertaining to the PFAM seed sequences (with incomplete information), need to be retrieved by the system from public databases. All necessary sequence data, primary structure and taxonomic information, is retrieved from either the NCBI database using extensible markup language (XML) or from the SWISSPROT database using its data communication interface. All protein sequences included by the user must be related by matching a common protein domain profile or by having significant similarity in their primary structure. Sequences in the gene set are aligned to a hidden Markov model (HMM) profile of a common protein domain. This profile-driven multiple alignment is performed by the hmmalign tool from the HMMER package (Eddy, 2001). Only matching states of the sequences are considered in the alignment. This approach includes only residues which effectively participate in the protein domain being analyzed, thus representing conserved regions. The use of matching states solely in the multiple sequence alignment renders a reliable dataset for the phylogenetic inference since these domains are more likely to be conserved in the evolutionary process. The underlying species phylogeny is inferred using ribosomal RNA data. The species sequence set contains aligned ribosomal sequences referring to the species parsed from the source organism fields in the gene sequence set. The ribosomal RNA sequences of the species list in the gene set are retrieved from the Ribosomal Database Project (RDP-II) (Cole et 4
5 al., 2003). The RDP-II is a repository that contains data from small subunit (SSU) ribosomal RNA of organisms from domains Eukaryota, Bacteria and Archaea. Ribosomal RNA sequences in the RDP-II databases are maintained at the repository in aligned format. These sequences are used for inferring the evolutionary history of the considered organisms and the domain must be specified in the RDP-II database system settings. The phylogenies of the gene set and the corresponding species set are inferred using the TREE-PUZZLE program package (Schmidt et al., 2002), which uses quartet-puzzling maximum likelihood phylogenetic analysis, including gamma distributed rate heterogeneity. The use of gamma distribution is computationally intensive and can be disabled in PGA if processing time is a limitation. The number of distinct rates and the alpha parameter should also be specified by the user. The phylogenetic inference of both sets renders polytomic unrooted trees. Gene duplication events are predicted using a methodology previously described (Zmasek and Eddy, 2001), modified to suit polytomic trees. Phylogenies of the gene and species sequence sets are presented in a single window, allowing the visual comparison of both trees. Predicted duplication events are depicted in the gene tree. IMPLEMENTATION PGA was written in C++ programming language (GNU gcc version 2.96) using Qt, a multi platform graphic user interface (GUI) application framework. Application data, such as sequence sets and the PFAM domain profiles are stored in a relational database based on a MySQL database management system. The tool was developed primarily for use in Intel based Linux workstations and can easily be ported to other platforms. An example is given in Fig. 1, where an Acethylcholine set is analysed. The main program window displays summary data the sequence set, the number of distinct species and PFAM profile used, in this case 7tm_1 (ACC: PF00001). The sequence set viewer shows the 5
6 profile-based multiple alignment and taxonomic information and the tree view displays the gene tree on the left and the species tree on the right. The potential gene duplication events (paralogy) are indicated by circles at the internodes. Selection of any sequence in either tree highlights all sequences originated from the same species (light gray in the figure and red in the actual application screen). Because the PGA maximum likelihood phylogenies are obtained by the quartet-puzzling methods the QP support, or the frequencies of branches in the space of possible quartets is presented in the final, maximum likelihood tree. Accordingly, QP values above 70% indicate potentially supported clusters and frequencies above 90% indicate strongly supported clusters. The QP scores can roughly be compared to Bootstrap values although should not be confused with. We recommend the TREE-PUZZLE documentation for a deeper discussion (Strimmer and von Haeseler, 1996). Acknowledgements P.B.P. received graduate scholarships from UNIFESP (Brazil). This work was supported by grants to M.R.S.B. from FAPESP and CNPq (Brazil), and the International Research Scholars Program of the Howard Hughes Medical Institute (USA). REFERENCES Bateman, A., Birney, E., Durbin, R., Eddy, S.R., Howe, K.L., Sonnhammer, E.L. (2000) The Pfam protein families database. Nucleic Acids Res., 28, Cole, J.R., Chai, B., Marsh, T.L., Farris, R.J., Wang, Q., Kulam, S.A., Chandra, S., McGarrell, D.M., Schmidt, T.M., Garrity, G.M., Tiedje, J.M. (2003) The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res., 31,
7 Eddy, S.R. (2001) HMMER: Profile hidden Markov models for biological sequence analysis ( Eisen, J.A. (1998) Phylogenomics: Improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res., 8, Schmidt, H.A., Strimmer, K., Vingron, M., von Haeseler, A. (2002) TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics, 18, Strimmer, K., and A. von Haeseler Quartet puzzling: a quartet maximum likelihood method for reconstructing tree topologies. Mol. Biol. Evol. 13 : Zmasek, C.M., Eddy, S.R. (2001) A simple algorithm to infer gene duplication and speciation events on a gene tree. Bioinformatics, 17,
8 Figure legend Fig. 1. PGA graphical user interface. The main program window displays summary data of an Acetylcholine sequence set, with the number of distinct species and PFAM profile used. The sequence set viewer shows the profile-based multiple alignment and taxonomic information. The tree view displays the sequence set tree on the left and the species tree on the right. Putative gene duplication events (paralogy) is indicated by circles. Selection of any sequence in the trees highlights all sequences originated from the same species (light gray in the figure and red in the actual application screen). Numbers above branch clusters indicate the quartet puzzling (QP support), where values above 70% indicate potentially supported clusters and frequencies above 90% indicate strongly supported clusters. The QP scores can roughly be compared to Bootstrap values although should not be confused with, please see TREE-PUZZLE documentation for a deeper discussion. 8
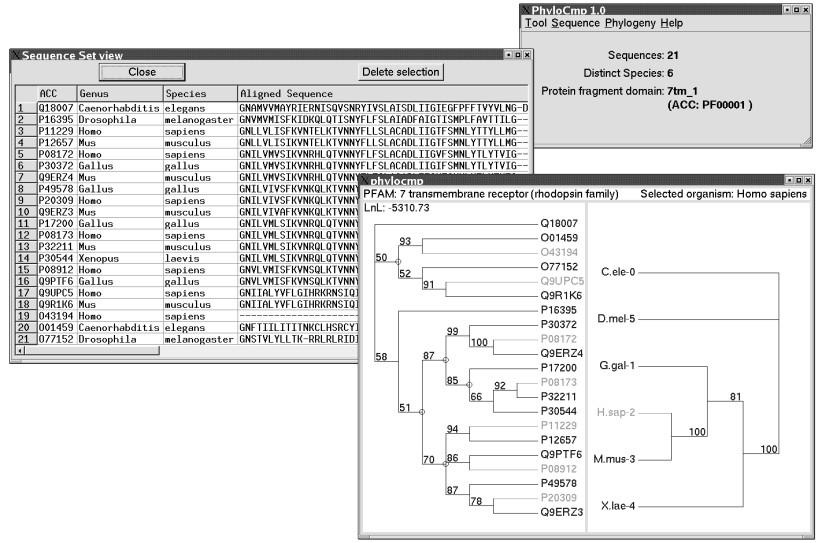
9 Fig. 1. 9
Phylogenetic relationship among S. castellii, S. cerevisiae and C. glabrata.
 Supplementary Note S2 Phylogenetic relationship among S. castellii, S. cerevisiae and C. glabrata. Phylogenetic trees reconstructed by a variety of methods from either single-copy orthologous loci (Class
Supplementary Note S2 Phylogenetic relationship among S. castellii, S. cerevisiae and C. glabrata. Phylogenetic trees reconstructed by a variety of methods from either single-copy orthologous loci (Class
Pipelining RDP Data to the Taxomatic Background Accomplishments vs objectives
 Pipelining RDP Data to the Taxomatic Timothy G. Lilburn, PI/Co-PI George M. Garrity, PI/Co-PI (Collaborative) James R. Cole, Co-PI (Collaborative) Project ID 0010734 Grant No. DE-FG02-04ER63932 Background
Pipelining RDP Data to the Taxomatic Timothy G. Lilburn, PI/Co-PI George M. Garrity, PI/Co-PI (Collaborative) James R. Cole, Co-PI (Collaborative) Project ID 0010734 Grant No. DE-FG02-04ER63932 Background
Bioinformatics tools for phylogeny and visualization. Yanbin Yin
 Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
SUPPLEMENTARY INFORMATION
 Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Session 5: Phylogenomics
 Session 5: Phylogenomics B.- Phylogeny based orthology assignment REMINDER: Gene tree reconstruction is divided in three steps: homology search, multiple sequence alignment and model selection plus tree
Session 5: Phylogenomics B.- Phylogeny based orthology assignment REMINDER: Gene tree reconstruction is divided in three steps: homology search, multiple sequence alignment and model selection plus tree
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut
 Faculty of Veterinary Medicine Assiut") Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
Elements of Bioinformatics 14F01 TP5 -Phylogenetic analysis
 Elements of Bioinformatics 14F01 TP5 -Phylogenetic analysis 10 December 2012 - Corrections - Exercise 1 Non-vertebrate chordates generally possess 2 homologs, vertebrates 3 or more gene copies; a Drosophila
Elements of Bioinformatics 14F01 TP5 -Phylogenetic analysis 10 December 2012 - Corrections - Exercise 1 Non-vertebrate chordates generally possess 2 homologs, vertebrates 3 or more gene copies; a Drosophila
Comparative Genomics II
 Comparative Genomics II Advances in Bioinformatics and Genomics GEN 240B Jason Stajich May 19 Comparative Genomics II Slide 1/31 Outline Introduction Gene Families Pairwise Methods Phylogenetic Methods
Comparative Genomics II Advances in Bioinformatics and Genomics GEN 240B Jason Stajich May 19 Comparative Genomics II Slide 1/31 Outline Introduction Gene Families Pairwise Methods Phylogenetic Methods
Chapter 26: Phylogeny and the Tree of Life Phylogenies Show Evolutionary Relationships
 Chapter 26: Phylogeny and the Tree of Life You Must Know The taxonomic categories and how they indicate relatedness. How systematics is used to develop phylogenetic trees. How to construct a phylogenetic
Chapter 26: Phylogeny and the Tree of Life You Must Know The taxonomic categories and how they indicate relatedness. How systematics is used to develop phylogenetic trees. How to construct a phylogenetic
Biology 559R: Introduction to Phylogenetic Comparative Methods Topics for this week:
 Biology 559R: Introduction to Phylogenetic Comparative Methods Topics for this week: Course general information About the course Course objectives Comparative methods: An overview R as language: uses and
Biology 559R: Introduction to Phylogenetic Comparative Methods Topics for this week: Course general information About the course Course objectives Comparative methods: An overview R as language: uses and
SUPPLEMENTARY INFORMATION
 Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Automating the Quest for Novel Prokaryotic Diversity (Revisited)
") Automating the Quest for Novel Prokaryotic Diversity (Revisited) 1,6 George M. Garrity, 5 Timothy G. Lilburn, 1 Scott H. Harrison, 2 Yun Bai, 3 Yuan Zhang, 6 Catherine Lyons and 4,6 James, R. Cole 1 Department
Automating the Quest for Novel Prokaryotic Diversity (Revisited) 1,6 George M. Garrity, 5 Timothy G. Lilburn, 1 Scott H. Harrison, 2 Yun Bai, 3 Yuan Zhang, 6 Catherine Lyons and 4,6 James, R. Cole 1 Department
Phylogenetic molecular function annotation
 Phylogenetic molecular function annotation Barbara E Engelhardt 1,1, Michael I Jordan 1,2, Susanna T Repo 3 and Steven E Brenner 3,4,2 1 EECS Department, University of California, Berkeley, CA, USA. 2
Phylogenetic molecular function annotation Barbara E Engelhardt 1,1, Michael I Jordan 1,2, Susanna T Repo 3 and Steven E Brenner 3,4,2 1 EECS Department, University of California, Berkeley, CA, USA. 2
 Tree of Life iological Sequence nalysis Chapter http://tolweb.org/tree/ Phylogenetic Prediction ll organisms on Earth have a common ancestor. ll species are related. The relationship is called a phylogeny
Tree of Life iological Sequence nalysis Chapter http://tolweb.org/tree/ Phylogenetic Prediction ll organisms on Earth have a common ancestor. ll species are related. The relationship is called a phylogeny
COMPUTING LARGE PHYLOGENIES WITH STATISTICAL METHODS: PROBLEMS & SOLUTIONS
 COMPUTING LARGE PHYLOGENIES WITH STATISTICAL METHODS: PROBLEMS & SOLUTIONS *Stamatakis A.P., Ludwig T., Meier H. Department of Computer Science, Technische Universität München Department of Computer Science,
COMPUTING LARGE PHYLOGENIES WITH STATISTICAL METHODS: PROBLEMS & SOLUTIONS *Stamatakis A.P., Ludwig T., Meier H. Department of Computer Science, Technische Universität München Department of Computer Science,
BINF6201/8201. Molecular phylogenetic methods
 BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction. Lesser Tenrec (Echinops telfairi)
") Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction Lesser Tenrec (Echinops telfairi) Goals: 1. Use phylogenetic experimental design theory to select optimal taxa to
Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction Lesser Tenrec (Echinops telfairi) Goals: 1. Use phylogenetic experimental design theory to select optimal taxa to
Basic Local Alignment Search Tool
 Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Phylogenetics: Building Phylogenetic Trees. COMP Fall 2010 Luay Nakhleh, Rice University
 Phylogenetics: Building Phylogenetic Trees COMP 571 - Fall 2010 Luay Nakhleh, Rice University Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary
Phylogenetics: Building Phylogenetic Trees COMP 571 - Fall 2010 Luay Nakhleh, Rice University Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary
A bioinformatics approach to the structural and functional analysis of the glycogen phosphorylase protein family
 A bioinformatics approach to the structural and functional analysis of the glycogen phosphorylase protein family Jieming Shen 1,2 and Hugh B. Nicholas, Jr. 3 1 Bioengineering and Bioinformatics Summer
A bioinformatics approach to the structural and functional analysis of the glycogen phosphorylase protein family Jieming Shen 1,2 and Hugh B. Nicholas, Jr. 3 1 Bioengineering and Bioinformatics Summer
Phylogenetics: Building Phylogenetic Trees
 1 Phylogenetics: Building Phylogenetic Trees COMP 571 Luay Nakhleh, Rice University 2 Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary model should
1 Phylogenetics: Building Phylogenetic Trees COMP 571 Luay Nakhleh, Rice University 2 Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary model should
Comparing whole genomes
 BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
Homology. and. Information Gathering and Domain Annotation for Proteins
 Homology and Information Gathering and Domain Annotation for Proteins Outline WHAT IS HOMOLOGY? HOW TO GATHER KNOWN PROTEIN INFORMATION? HOW TO ANNOTATE PROTEIN DOMAINS? EXAMPLES AND EXERCISES Homology
Homology and Information Gathering and Domain Annotation for Proteins Outline WHAT IS HOMOLOGY? HOW TO GATHER KNOWN PROTEIN INFORMATION? HOW TO ANNOTATE PROTEIN DOMAINS? EXAMPLES AND EXERCISES Homology
BLAST. Varieties of BLAST
 BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
Dr. Amira A. AL-Hosary
 Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
MiGA: The Microbial Genome Atlas
 December 12 th 2017 MiGA: The Microbial Genome Atlas Jim Cole Center for Microbial Ecology Dept. of Plant, Soil & Microbial Sciences Michigan State University East Lansing, Michigan U.S.A. Where I m From
December 12 th 2017 MiGA: The Microbial Genome Atlas Jim Cole Center for Microbial Ecology Dept. of Plant, Soil & Microbial Sciences Michigan State University East Lansing, Michigan U.S.A. Where I m From
Comparison of Three Fugal ITS Reference Sets. Qiong Wang and Jim R. Cole
 RDP TECHNICAL REPORT Created 04/12/2014, Updated 08/08/2014 Summary Comparison of Three Fugal ITS Reference Sets Qiong Wang and Jim R. Cole wangqion@msu.edu, colej@msu.edu In this report, we evaluate the
RDP TECHNICAL REPORT Created 04/12/2014, Updated 08/08/2014 Summary Comparison of Three Fugal ITS Reference Sets Qiong Wang and Jim R. Cole wangqion@msu.edu, colej@msu.edu In this report, we evaluate the
BLAST Database Searching. BME 110: CompBio Tools Todd Lowe April 8, 2010
 BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
Mitochondrial Genome Annotation
 Protein Genes 1,2 1 Institute of Bioinformatics University of Leipzig 2 Department of Bioinformatics Lebanese University TBI Bled 2015 Outline Introduction Mitochondrial DNA Problem Tools Training Annotation
Protein Genes 1,2 1 Institute of Bioinformatics University of Leipzig 2 Department of Bioinformatics Lebanese University TBI Bled 2015 Outline Introduction Mitochondrial DNA Problem Tools Training Annotation
Inferring phylogeny. Constructing phylogenetic trees. Tõnu Margus. Bioinformatics MTAT
 Inferring phylogeny Constructing phylogenetic trees Tõnu Margus Contents What is phylogeny? How/why it is possible to infer it? Representing evolutionary relationships on trees What type questions questions
Inferring phylogeny Constructing phylogenetic trees Tõnu Margus Contents What is phylogeny? How/why it is possible to infer it? Representing evolutionary relationships on trees What type questions questions
Research Proposal. Title: Multiple Sequence Alignment used to investigate the co-evolving positions in OxyR Protein family.
 Research Proposal Title: Multiple Sequence Alignment used to investigate the co-evolving positions in OxyR Protein family. Name: Minjal Pancholi Howard University Washington, DC. June 19, 2009 Research
Research Proposal Title: Multiple Sequence Alignment used to investigate the co-evolving positions in OxyR Protein family. Name: Minjal Pancholi Howard University Washington, DC. June 19, 2009 Research
Bioinformatics. Dept. of Computational Biology & Bioinformatics
 Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Homology and Information Gathering and Domain Annotation for Proteins
 Homology and Information Gathering and Domain Annotation for Proteins Outline Homology Information Gathering for Proteins Domain Annotation for Proteins Examples and exercises The concept of homology The
Homology and Information Gathering and Domain Annotation for Proteins Outline Homology Information Gathering for Proteins Domain Annotation for Proteins Examples and exercises The concept of homology The
Microbes usually have few distinguishing properties that relate them, so a hierarchical taxonomy mainly has not been possible.
 Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Microbial Taxonomy. Slowly evolving molecules (e.g., rrna) used for large-scale structure; "fast- clock" molecules for fine-structure.
 used for large-scale structure; fast- clock molecules for fine-structure.") Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Phylogenetic Tree Reconstruction
 I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
Bioinformatics. Proteins II. - Pattern, Profile, & Structure Database Searching. Robert Latek, Ph.D. Bioinformatics, Biocomputing
 Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Orthology Part I: concepts and implications Toni Gabaldón Centre for Genomic Regulation (CRG), Barcelona
, Barcelona") Orthology Part I: concepts and implications Toni Gabaldón Centre for Genomic Regulation (CRG), Barcelona (tgabaldon@crg.es) http://gabaldonlab.crg.es Homology the same organ in different animals under
Orthology Part I: concepts and implications Toni Gabaldón Centre for Genomic Regulation (CRG), Barcelona (tgabaldon@crg.es) http://gabaldonlab.crg.es Homology the same organ in different animals under
8/23/2014. Phylogeny and the Tree of Life
 Phylogeny and the Tree of Life Chapter 26 Objectives Explain the following characteristics of the Linnaean system of classification: a. binomial nomenclature b. hierarchical classification List the major
Phylogeny and the Tree of Life Chapter 26 Objectives Explain the following characteristics of the Linnaean system of classification: a. binomial nomenclature b. hierarchical classification List the major
Taxonomy. Content. How to determine & classify a species. Phylogeny and evolution
 Taxonomy Content Why Taxonomy? How to determine & classify a species Domains versus Kingdoms Phylogeny and evolution Why Taxonomy? Classification Arrangement in groups or taxa (taxon = group) Nomenclature
Taxonomy Content Why Taxonomy? How to determine & classify a species Domains versus Kingdoms Phylogeny and evolution Why Taxonomy? Classification Arrangement in groups or taxa (taxon = group) Nomenclature
CHAPTERS 24-25: Evidence for Evolution and Phylogeny
 CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Microbial Taxonomy and the Evolution of Diversity
 19 Microbial Taxonomy and the Evolution of Diversity Copyright McGraw-Hill Global Education Holdings, LLC. Permission required for reproduction or display. 1 Taxonomy Introduction to Microbial Taxonomy
19 Microbial Taxonomy and the Evolution of Diversity Copyright McGraw-Hill Global Education Holdings, LLC. Permission required for reproduction or display. 1 Taxonomy Introduction to Microbial Taxonomy
08/21/2017 BLAST. Multiple Sequence Alignments: Clustal Omega
 BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
reconciling trees Stefanie Hartmann postdoc, Todd Vision s lab University of North Carolina the data
 reconciling trees Stefanie Hartmann postdoc, Todd Vision s lab University of North Carolina 1 the data alignments and phylogenies for ~27,000 gene families from 140 plant species www.phytome.org publicly
reconciling trees Stefanie Hartmann postdoc, Todd Vision s lab University of North Carolina 1 the data alignments and phylogenies for ~27,000 gene families from 140 plant species www.phytome.org publicly
Molecular phylogeny How to infer phylogenetic trees using molecular sequences
 Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 200 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 200 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Introduction to Evolutionary Concepts
 Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2009 VMD/MultiSeq
Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2009 VMD/MultiSeq
CISC 636 Computational Biology & Bioinformatics (Fall 2016)
") CISC 636 Computational Biology & Bioinformatics (Fall 2016) Predicting Protein-Protein Interactions CISC636, F16, Lec22, Liao 1 Background Proteins do not function as isolated entities. Protein-Protein
CISC 636 Computational Biology & Bioinformatics (Fall 2016) Predicting Protein-Protein Interactions CISC636, F16, Lec22, Liao 1 Background Proteins do not function as isolated entities. Protein-Protein
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Distance Methods Character Methods
Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Distance Methods Character Methods
Molecular phylogeny How to infer phylogenetic trees using molecular sequences
 Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 2009 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 2009 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Subfamily HMMS in Functional Genomics. D. Brown, N. Krishnamurthy, J.M. Dale, W. Christopher, and K. Sjölander
 Subfamily HMMS in Functional Genomics D. Brown, N. Krishnamurthy, J.M. Dale, W. Christopher, and K. Sjölander Pacific Symposium on Biocomputing 10:322-333(2005) SUBFAMILY HMMS IN FUNCTIONAL GENOMICS DUNCAN
Subfamily HMMS in Functional Genomics D. Brown, N. Krishnamurthy, J.M. Dale, W. Christopher, and K. Sjölander Pacific Symposium on Biocomputing 10:322-333(2005) SUBFAMILY HMMS IN FUNCTIONAL GENOMICS DUNCAN
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Title ghost-tree: creating hybrid-gene phylogenetic trees for diversity analyses
 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 Title ghost-tree: creating hybrid-gene phylogenetic trees for diversity analyses
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 Title ghost-tree: creating hybrid-gene phylogenetic trees for diversity analyses
Genomic insights into the taxonomic status of the Bacillus cereus group. Laboratory of Marine Genetic Resources, Third Institute of Oceanography,
 1 2 3 Genomic insights into the taxonomic status of the Bacillus cereus group Yang Liu 1, Qiliang Lai 1, Markus Göker 2, Jan P. Meier-Kolthoff 2, Meng Wang 3, Yamin Sun 3, Lei Wang 3 and Zongze Shao 1*
1 2 3 Genomic insights into the taxonomic status of the Bacillus cereus group Yang Liu 1, Qiliang Lai 1, Markus Göker 2, Jan P. Meier-Kolthoff 2, Meng Wang 3, Yamin Sun 3, Lei Wang 3 and Zongze Shao 1*
Measuring quaternary structure similarity using global versus local measures.
 Supplementary Figure 1 Measuring quaternary structure similarity using global versus local measures. (a) Structural similarity of two protein complexes can be inferred from a global superposition, which
Supplementary Figure 1 Measuring quaternary structure similarity using global versus local measures. (a) Structural similarity of two protein complexes can be inferred from a global superposition, which
Introduction to protein alignments
 Introduction to protein alignments Comparative Analysis of Proteins Experimental evidence from one or more proteins can be used to infer function of related protein(s). Gene A Gene X Protein A compare
Introduction to protein alignments Comparative Analysis of Proteins Experimental evidence from one or more proteins can be used to infer function of related protein(s). Gene A Gene X Protein A compare
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Supplementary material to Whitney, K. D., B. Boussau, E. J. Baack, and T. Garland Jr. in press. Drift and genome complexity revisited. PLoS Genetics.
 Supplementary material to Whitney, K. D., B. Boussau, E. J. Baack, and T. Garland Jr. in press. Drift and genome complexity revisited. PLoS Genetics. Tree topologies Two topologies were examined, one favoring
Supplementary material to Whitney, K. D., B. Boussau, E. J. Baack, and T. Garland Jr. in press. Drift and genome complexity revisited. PLoS Genetics. Tree topologies Two topologies were examined, one favoring
The Phylogenetic Handbook
 The Phylogenetic Handbook A Practical Approach to DNA and Protein Phylogeny Edited by Marco Salemi University of California, Irvine and Katholieke Universiteit Leuven, Belgium and Anne-Mieke Vandamme Rega
The Phylogenetic Handbook A Practical Approach to DNA and Protein Phylogeny Edited by Marco Salemi University of California, Irvine and Katholieke Universiteit Leuven, Belgium and Anne-Mieke Vandamme Rega
Introduction to Bioinformatics Online Course: IBT
 Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
"Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky
 MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
Gene function annotation
 Gene function annotation Paul D. Thomas, Ph.D. University of Southern California What is function annotation? The formal answer to the question: what does this gene do? The association between: a description
Gene function annotation Paul D. Thomas, Ph.D. University of Southern California What is function annotation? The formal answer to the question: what does this gene do? The association between: a description
Computational approaches for functional genomics
 Computational approaches for functional genomics Kalin Vetsigian October 31, 2001 The rapidly increasing number of completely sequenced genomes have stimulated the development of new methods for finding
Computational approaches for functional genomics Kalin Vetsigian October 31, 2001 The rapidly increasing number of completely sequenced genomes have stimulated the development of new methods for finding
9/30/11. Evolution theory. Phylogenetic Tree Reconstruction. Phylogenetic trees (binary trees) Phylogeny (phylogenetic tree)
 Phylogeny (phylogenetic tree)") I9 Introduction to Bioinformatics, 0 Phylogenetic ree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & omputing, IUB Evolution theory Speciation Evolution of new organisms is driven by
I9 Introduction to Bioinformatics, 0 Phylogenetic ree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & omputing, IUB Evolution theory Speciation Evolution of new organisms is driven by
Supplementary Materials for
 advances.sciencemag.org/cgi/content/full/1/8/e1500527/dc1 Supplementary Materials for A phylogenomic data-driven exploration of viral origins and evolution The PDF file includes: Arshan Nasir and Gustavo
advances.sciencemag.org/cgi/content/full/1/8/e1500527/dc1 Supplementary Materials for A phylogenomic data-driven exploration of viral origins and evolution The PDF file includes: Arshan Nasir and Gustavo
Introduction to Bioinformatics
 Introduction to Bioinformatics Lecture : p he biological problem p lobal alignment p Local alignment p Multiple alignment 6 Background: comparative genomics p Basic question in biology: what properties
Introduction to Bioinformatics Lecture : p he biological problem p lobal alignment p Local alignment p Multiple alignment 6 Background: comparative genomics p Basic question in biology: what properties
UoN, CAS, DBSC BIOL102 lecture notes by: Dr. Mustafa A. Mansi. The Phylogenetic Systematics (Phylogeny and Systematics)
") - Phylogeny? - Systematics? The Phylogenetic Systematics (Phylogeny and Systematics) - Phylogenetic systematics? Connection between phylogeny and classification. - Phylogenetic systematics informs the
- Phylogeny? - Systematics? The Phylogenetic Systematics (Phylogeny and Systematics) - Phylogenetic systematics? Connection between phylogeny and classification. - Phylogenetic systematics informs the
Unsupervised Learning in Spectral Genome Analysis
 Unsupervised Learning in Spectral Genome Analysis Lutz Hamel 1, Neha Nahar 1, Maria S. Poptsova 2, Olga Zhaxybayeva 3, J. Peter Gogarten 2 1 Department of Computer Sciences and Statistics, University of
Unsupervised Learning in Spectral Genome Analysis Lutz Hamel 1, Neha Nahar 1, Maria S. Poptsova 2, Olga Zhaxybayeva 3, J. Peter Gogarten 2 1 Department of Computer Sciences and Statistics, University of
Supplementary text for the section Interactions conserved across species: can one select the conserved interactions?
 1 Supporting Information: What Evidence is There for the Homology of Protein-Protein Interactions? Anna C. F. Lewis, Nick S. Jones, Mason A. Porter, Charlotte M. Deane Supplementary text for the section
1 Supporting Information: What Evidence is There for the Homology of Protein-Protein Interactions? Anna C. F. Lewis, Nick S. Jones, Mason A. Porter, Charlotte M. Deane Supplementary text for the section
Bioinformatics Exercises
 Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
Hands-On Nine The PAX6 Gene and Protein
 Hands-On Nine The PAX6 Gene and Protein Main Purpose of Hands-On Activity: Using bioinformatics tools to examine the sequences, homology, and disease relevance of the Pax6: a master gene of eye formation.
Hands-On Nine The PAX6 Gene and Protein Main Purpose of Hands-On Activity: Using bioinformatics tools to examine the sequences, homology, and disease relevance of the Pax6: a master gene of eye formation.
A (short) introduction to phylogenetics
 introduction to phylogenetics") A (short) introduction to phylogenetics Thibaut Jombart, Marie-Pauline Beugin MRC Centre for Outbreak Analysis and Modelling Imperial College London Genetic data analysis with PR Statistics, Millport Field
A (short) introduction to phylogenetics Thibaut Jombart, Marie-Pauline Beugin MRC Centre for Outbreak Analysis and Modelling Imperial College London Genetic data analysis with PR Statistics, Millport Field
Phylogenetics. Applications of phylogenetics. Unrooted networks vs. rooted trees. Outline
 Phylogenetics Todd Vision iology 522 March 26, 2007 pplications of phylogenetics Studying organismal or biogeographic history Systematics ating events in the fossil record onservation biology Studying
Phylogenetics Todd Vision iology 522 March 26, 2007 pplications of phylogenetics Studying organismal or biogeographic history Systematics ating events in the fossil record onservation biology Studying
Consensus Methods. * You are only responsible for the first two
 Consensus Trees * consensus trees reconcile clades from different trees * consensus is a conservative estimate of phylogeny that emphasizes points of agreement * philosophy: agreement among data sets is
Consensus Trees * consensus trees reconcile clades from different trees * consensus is a conservative estimate of phylogeny that emphasizes points of agreement * philosophy: agreement among data sets is
CS612 - Algorithms in Bioinformatics
 Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
SI Materials and Methods
 SI Materials and Methods Gibbs Sampling with Informative Priors. Full description of the PhyloGibbs algorithm, including comprehensive tests on synthetic and yeast data sets, can be found in Siddharthan
SI Materials and Methods Gibbs Sampling with Informative Priors. Full description of the PhyloGibbs algorithm, including comprehensive tests on synthetic and yeast data sets, can be found in Siddharthan
Comparative Bioinformatics Midterm II Fall 2004
 Comparative Bioinformatics Midterm II Fall 2004 Objective Answer, part I: For each of the following, select the single best answer or completion of the phrase. (3 points each) 1. Deinococcus radiodurans
Comparative Bioinformatics Midterm II Fall 2004 Objective Answer, part I: For each of the following, select the single best answer or completion of the phrase. (3 points each) 1. Deinococcus radiodurans
Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics
 Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics Tandy Warnow Founder Professor of Engineering The University of Illinois at Urbana-Champaign http://tandy.cs.illinois.edu
Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics Tandy Warnow Founder Professor of Engineering The University of Illinois at Urbana-Champaign http://tandy.cs.illinois.edu
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Quartet Inference from SNP Data Under the Coalescent Model
 Bioinformatics Advance Access published August 7, 2014 Quartet Inference from SNP Data Under the Coalescent Model Julia Chifman 1 and Laura Kubatko 2,3 1 Department of Cancer Biology, Wake Forest School
Bioinformatics Advance Access published August 7, 2014 Quartet Inference from SNP Data Under the Coalescent Model Julia Chifman 1 and Laura Kubatko 2,3 1 Department of Cancer Biology, Wake Forest School
Effects of Gap Open and Gap Extension Penalties
 Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Microbiome: 16S rrna Sequencing 3/30/2018
 Microbiome: 16S rrna Sequencing 3/30/2018 Skills from Previous Lectures Central Dogma of Biology Lecture 3: Genetics and Genomics Lecture 4: Microarrays Lecture 12: ChIP-Seq Phylogenetics Lecture 13: Phylogenetics
Microbiome: 16S rrna Sequencing 3/30/2018 Skills from Previous Lectures Central Dogma of Biology Lecture 3: Genetics and Genomics Lecture 4: Microarrays Lecture 12: ChIP-Seq Phylogenetics Lecture 13: Phylogenetics
Multiple sequence alignment
 Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
PSODA: Better Tasting and Less Filling Than PAUP
 Brigham Young University BYU ScholarsArchive All Faculty Publications 2007-10-01 PSODA: Better Tasting and Less Filling Than PAUP Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu
Brigham Young University BYU ScholarsArchive All Faculty Publications 2007-10-01 PSODA: Better Tasting and Less Filling Than PAUP Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu
Cluster Analysis of Gene Expression Microarray Data. BIOL 495S/ CS 490B/ MATH 490B/ STAT 490B Introduction to Bioinformatics April 8, 2002
 Cluster Analysis of Gene Expression Microarray Data BIOL 495S/ CS 490B/ MATH 490B/ STAT 490B Introduction to Bioinformatics April 8, 2002 1 Data representations Data are relative measurements log 2 ( red
Cluster Analysis of Gene Expression Microarray Data BIOL 495S/ CS 490B/ MATH 490B/ STAT 490B Introduction to Bioinformatics April 8, 2002 1 Data representations Data are relative measurements log 2 ( red
Supplementary Information
 Supplementary Information Supplementary Figure 1. Schematic pipeline for single-cell genome assembly, cleaning and annotation. a. The assembly process was optimized to account for multiple cells putatively
Supplementary Information Supplementary Figure 1. Schematic pipeline for single-cell genome assembly, cleaning and annotation. a. The assembly process was optimized to account for multiple cells putatively
1 Abstract. 2 Introduction. 3 Requirements. 4 Procedure
 1 Abstract None 2 Introduction The archaeal core set is used in testing the completeness of the archaeal draft genomes. The core set comprises of conserved single copy genes from 25 genomes. Coverage statistic
1 Abstract None 2 Introduction The archaeal core set is used in testing the completeness of the archaeal draft genomes. The core set comprises of conserved single copy genes from 25 genomes. Coverage statistic
Part III - Bioinformatics Study of Aminoacyl trna Synthetases. VMD Multiseq Tutorial Web tools. Perth, Australia 2004 Computational Biology Workshop
 Part III - Bioinformatics Study of Aminoacyl trna Synthetases VMD Multiseq Tutorial Web tools Perth, Australia 2004 Computational Biology Workshop Multiple Sequence Alignments The aminoacyl-trna synthetases,
Part III - Bioinformatics Study of Aminoacyl trna Synthetases VMD Multiseq Tutorial Web tools Perth, Australia 2004 Computational Biology Workshop Multiple Sequence Alignments The aminoacyl-trna synthetases,
Bio 1B Lecture Outline (please print and bring along) Fall, 2007
 Fall, 2007") Bio 1B Lecture Outline (please print and bring along) Fall, 2007 B.D. Mishler, Dept. of Integrative Biology 2-6810, bmishler@berkeley.edu Evolution lecture #5 -- Molecular genetics and molecular evolution
Bio 1B Lecture Outline (please print and bring along) Fall, 2007 B.D. Mishler, Dept. of Integrative Biology 2-6810, bmishler@berkeley.edu Evolution lecture #5 -- Molecular genetics and molecular evolution
SnoPatrol: How many snorna genes are there? Supplementary
 SnoPatrol: How many snorna genes are there? Supplementary materials. Paul P. Gardner 1, Alex G. Bateman 1 and Anthony M. Poole 2,3 1 Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton,
SnoPatrol: How many snorna genes are there? Supplementary materials. Paul P. Gardner 1, Alex G. Bateman 1 and Anthony M. Poole 2,3 1 Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton,
Microbial Taxonomy. Microbes usually have few distinguishing properties that relate them, so a hierarchical taxonomy mainly has not been possible.
 Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
An Automated Phylogenetic Tree-Based Small Subunit rrna Taxonomy and Alignment Pipeline (STAP)
") An Automated Phylogenetic Tree-Based Small Subunit rrna Taxonomy and Alignment Pipeline (STAP) Dongying Wu 1 *, Amber Hartman 1,6, Naomi Ward 4,5, Jonathan A. Eisen 1,2,3 1 UC Davis Genome Center, University
An Automated Phylogenetic Tree-Based Small Subunit rrna Taxonomy and Alignment Pipeline (STAP) Dongying Wu 1 *, Amber Hartman 1,6, Naomi Ward 4,5, Jonathan A. Eisen 1,2,3 1 UC Davis Genome Center, University
Biology 559R: Introduction to Phylogenetic Comparative Methods Topics for this week (Jan 27 & 29):
:") Biology 559R: Introduction to Phylogenetic Comparative Methods Topics for this week (Jan 27 & 29): Statistical estimation of models of sequence evolution Phylogenetic inference using maximum likelihood:
Biology 559R: Introduction to Phylogenetic Comparative Methods Topics for this week (Jan 27 & 29): Statistical estimation of models of sequence evolution Phylogenetic inference using maximum likelihood:
Evolutionary Rate Covariation of Domain Families
 Evolutionary Rate Covariation of Domain Families Author: Brandon Jernigan A Thesis Submitted to the Department of Chemistry and Biochemistry in Partial Fulfillment of the Bachelors of Science Degree in
Evolutionary Rate Covariation of Domain Families Author: Brandon Jernigan A Thesis Submitted to the Department of Chemistry and Biochemistry in Partial Fulfillment of the Bachelors of Science Degree in
Sequence alignment methods. Pairwise alignment. The universe of biological sequence analysis
 he universe of biological sequence analysis Word/pattern recognition- Identification of restriction enzyme cleavage sites Sequence alignment methods PstI he universe of biological sequence analysis - prediction
he universe of biological sequence analysis Word/pattern recognition- Identification of restriction enzyme cleavage sites Sequence alignment methods PstI he universe of biological sequence analysis - prediction
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Orthology Part I concepts and implications Toni Gabaldón Centre for Genomic Regulation (CRG), Barcelona
, Barcelona") Orthology Part I concepts and implications Toni Gabaldón Centre for Genomic Regulation (CRG), Barcelona Toni Gabaldón Contact: tgabaldon@crg.es Group website: http://gabaldonlab.crg.es Science blog: http://treevolution.blogspot.com
Orthology Part I concepts and implications Toni Gabaldón Centre for Genomic Regulation (CRG), Barcelona Toni Gabaldón Contact: tgabaldon@crg.es Group website: http://gabaldonlab.crg.es Science blog: http://treevolution.blogspot.com