Homology. Bio5488 Ting Wang 1/25/15, 1/27/15
|
|
|
- Bruno Matthews
- 6 years ago
- Views:
Transcription
1 Homology Bio5488 Ting Wang 1/25/15, 1/27/15
2 ACGTTGCCACTTTCCGGGCCACCTGGCCACCTTATTTTCGGAAATATACCGGGCCTTTTTT x x CTTTCCCGGCCTCCTGGCCA match: +1 mismatch: -1 matching score = 16 How to align them? Why we can align them? Why +1 for match, and -1 for mismatch? What does the score mean? Is 16 a good score?
3 Outline Nobel-price-worthy work on homology What is homology? How to detect homology? How to quantify homology? How to use homology? Homology beyond sequence analysis Next-gen sequencing alignment


4 Russell Doolittle (Bishop and Varmus)
 What is the significance?")
5 Bishop and Varmus strategy (Nobel price 1989) Doolittle strategy (could be the first Nobel price for computational biology) What is the significance?
6 A few Definitions Homologs: genes/sequences sharing a common origin Orthologs: genes originating from a single ancestral gene in the last common ancestor of the compared genomes; genes related via speciation Paralogs: genes related via duplication Xenolog: sequences that have arisen out of horizontal transfer events (symbiosis, viruses, etc) Co-orthologs: two or more genes in one lineage that are, collectively, orthologous to one or more genes in another lineage due to a lineage-specific duplication(s) Outparalogs: paralogous genes resulting from a duplication(s) preceding a given speciation event Inparalogs: paralogous genes resulting from a lineage-specific duplication(s) subsequent to a given speciation event
7 Relation of sequences Need ancestral sequences to distinguish orthologs and paralogs (exercise on board)
8 Similarity versus Homology Similarity refers to the likeness or % identity between 2 sequences Similarity means sharing a statistically significant number of bases or amino acids Similarity does not imply homology Similarity can be quantified It is ok to say that two sequences are X% identical It is ok to say that two sequences have a similarity score of Z It is generally incorrect to say that two sequences are X% similar Homology refers to shared ancestry Two sequences are homologous if they are derived from a common ancestral sequence Homology usually implies similarity Low complexity regions can be highly similar without being homologous Homologous sequences are not always highly similar A sequence is either homologous or not. Never say two things are X% homologous
9 Why Compare Sequences? Sequence comparisons lie at the heart of all bioinformatics Identify sequences What is this thing I just found? Compare new genes to known ones Compare genes from different species information about evolution Guess functions for entire genomes full of new gene sequences Metagenomics What does it matter if two sequences are similar or not? Globally similar sequences are likely to have the same biological function or role Locally similar sequences are likely to have some physical shape or property with similar biochemical roles If we can figure out what one does, we may be able to figure out what they all do
10 Sequence alignment How to optimally align two sequences Dot plots Dynamic programming Global alignment Local alignment How to score an alignment Fast similar sequence search BLAST BLAT More recent development: short read alignment Determine statistical significance Using information in multiple sequence alignment to improve sensitivity
11 Visual Alignments (Dot Plots) Build a comparison matrix Rows: Sequence #1 Columns: Sequence #2 Filling For each coordinate, if the character in the row matches the one in the column, fill in the cell Continue until all coordinates have been examined

12 Example Dot Plot
13 Noise in Dot Plots Nucleic Acids (DNA, RNA) 1 out of 4 bases matches at random Windowing helps reduce noise Can require >X bp match before plotting Percentage of bases matching in the window is set as threshold
14 Met14 vs Met2 DotPlot MET14 (1000nt) Match = 1 Mismatch =-1 Gray: 1
15 Met14 vs Met2 MET14 (1000nt) Red: >5
16 chain of human hemoglobin chain of human hemoglobin

17 MAZ: Myc associated zinc finger isoform 1 self alignment

18 Human vs Chimp Y chromosome comparison
19 Dot plots of DNA sequence identity between chimpanzee and human Y chromosomes and chromosomes 21. JF Hughes et al. Nature 000, 1-4 (2010) doi: /nature08700
20 Aligning sequences by residue Match: award Mismatch (substitution or mutation): penalize Insertion/Deletion (INDELS gaps): penalize (gap open, gap extension) A L I G N M E N T - L I G A M E N T
21 More than one solution is possible Which alignment is best? A T C G G A T - C T A C G G A C T A T C G G A T C T A C G G A C T
22 Alignment Scoring Scheme Possible scoring scheme: match: +2 mismatch: -1 indel 2 Alignment 1: 5*2 + 1*-1 + 4*-2 = = 1 Alignment 2: 6*2 + 1*-1 + 2*-2 = = 7
23 Dynamic Programming Global Alignments: Needleman S.B. and Wunsch C.D. (1970) J. Mol. Biol. 48, Local Alignments: Smith T.F. and Waterman M.S. (1981) J. Mol. Biol.147, One simple modification of Needleman/Wunsch: when a value in the score matrix becomes negative, reset it to zero (begin of new alignment) Guaranteed to be mathematically optimal: Given two sequences (and a scoring system) these algorithms are guaranteed to find the very best alignment between the two sequences! Slow N 2 algorithm Performed in 2 stages Prepare a scoring matrix using recursive function Scan matrix diagonally using traceback protocol
24
25 Dynamic Programming G E N E T I C S G E N E S I S G E N E T I C S G E N E S I S G E N E T I C S * G E N E S I S
26 DP (demo) Match=5, mismatch=-3, gap=-4
27 DP (demo) S 1,1 = MAX{S 0,0 + 5, S 1,0-4, S 0,1 4,0} = MAX{5, -4, -4, 0} = 5
28 DP (demo) S 1,2 = MAX{S 0,1-3, S 1,1-4, S 0,2 4, 0} = MAX{0-3, 5 4, 0 4, 0} = MAX{-3, 1, -4, 0} = 1
29 DP (demo) S 1,3 = MAX{S 0,2-3, S 1,2-4, S 0,3 4, 0} = MAX{0-3, 1 4, 0 4, 0} = MAX{-3, -3, -4, 0} = 0
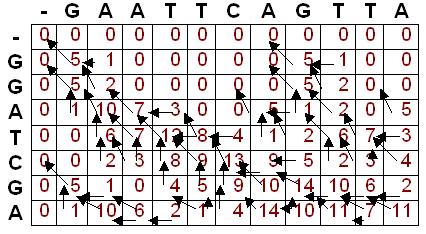
30 DP (demo)
31 Trace Back (Local Alignment) Maximum local alignment score is the highest score anywhere in the matrix (14 in this example) 14 is found in two separate cells, indicating two possible multiple alignments producing the maximal local alignment score
32 Trace Back (Local Alignment) Traceback begins in the position with the highest value. At each cell, we look to see where we move next according to the pointers When a cell is reached where there is not a pointer to a previous cell, we have reached the beginning of the alignment

33 Trace Back Demo

34 Trace Back Demo

35 Trace Back Demo
36 Maximum Local Alignment G A A T T C - A G G A T C G A G A A T T C - A G G A T C G A = =14
37 Linear vs. Affine Gaps So far, gaps have been modeled as linear More likely contiguous block of residues inserted or deleted 1 gap of length k rather than k gaps of length 1 Can create scoring scheme to penalize big gaps relatively less Biggest cost is to open new gap, but extending is not so costly
38 Affine Gap Penalty w x = g + r(x-1) w x : total gap penalty g: gap open penalty r: gap extend penalty x: gap length gap penalty chosen relative to score matrix
39 Scoring Alignments Pick a scoring matrix BLOSUM62 PAM250 Match=5, mismatch=-4 Decide on gap penalties -gap opening penalty (-8) -gap extension penalty (-1) Assume every position is independent Sum scores at each position [log(x*y)=logx+logy]
40 Scoring Matrices S ij = log( q ij p i p j ) An empirical model of evolution, biology and chemistry all wrapped up in a 20 X 20 (or 4 X 4) table of numbers Structurally or chemically similar residues should ideally have high diagonal or off-diagonal numbers Structurally or chemically dissimilar residues should ideally have low diagonal or off-diagonal numbers What does the score mean: The likelihood of seeing two residues align (preserved) than random expected. l
41 Scoring Alignments Blosum62 Scoring Matrix
42 BLOSUM substitution matrices Developed for distantly related proteins Substitutions only from multiple alignments of conserved regions of protein families, hand curated, constitute the known homologous blocks Identity threshold to define conserved blocks can be varied, e.g. 62% idenitity gives BLOSUM62 Scores calculated from frequency of amino acids in aligned pairs compared to what would be expected due to abundance alone, given all sequences Henikoff and Henikoff (1992) Proc. Natl. Acad. Sci. USA 89,
43 Blosum Matricies What score should we give to a ser residue aligned with a thr residue? score(s: T) log2 P(S: T homology) P(S: T random)
44 example of deriving Blosum scores for S:S, S:T, and T:T Database of known alignments SDHIP HKSA WMFET RTQC SDHLP HRTA WMFDT RTNC SDHIP HKSG WLFDT KTQC SEHLP KSQC SEHLP KTQC Homology Model (consider each pair of sequences separately) S:S pairs in alignments = 11 P(S:S homology) = 11/117 =.094 S:T pairs in alignments = 6 P(S:T homology) = 6/117 =.051 T:T pairs in alignments = 9 P(T:T homology) = 9/117 =.078 Total pairs in alignments = 117
45 example of deriving Blosum scores for S:S, S:T, and T:T Database of known alignments SDHIP HKSA WMFET RTQC SDHLP HRTA WMFDT RTNC SDHIP HKSG WLFDT KTQC SEHLP KSQC SEHLP KTQC Random Model Number of S residues = 8 P(S:S random)=p(s)p(s)=(8/72) 2 =.012 Number of T residues = 8 P(S:T random)=2*p(s)p(t)=2*(8/72) 2 =.024 Total residues = 72 P(T:T random)=p(t)p(t)=(8/72) 2 =.012
46 example of deriving Blosum scores for S:S, S:T, and T:T score(s: S) log 2 P(S:S homology) P(S:S random) log score(s: T) log 2 P(S: T homology) P(S: T random) log score(t: T) log 2 P(T : T homology) P(T : T random) log
47 BLOSUM and PAM BLOSUM 45 BLOSUM 62 BLOSUM 90 PAM 250 PAM 160 PAM 100 More Divergent Less Divergent BLOSUM 62 is the default matrix in BLAST 2.0. Though it is tailored for comparisons of moderately distant proteins, it performs well in detecting closer relationships. A search for distant relatives may be more sensitive with a different matrix. PAM matrices: point accepted mutation
48 Scoring Matrices Take Home Points Based on log odds scores Ratios>1 give positive scores, ratios<1 give negative scores Because log(x*y)=logx+logy the score of an alignment is the sum of the scores for each pair of aligned residues Assume independence of adjacent residues when scoring Introduced the concept that the frequency of a residue in a multiple alignment is informative
49 Fast Similar Sequence Search Can we run Smith-Waterman between query and every DB sequence? Yes, but too slow! General approach Break query and DB sequence to match subsequences Extend the matched subsequences, filter hopeless sequences Use dynamic programming to get optimal alignment
50 BLAST Basic Local Alignment Search Tool Altschul et al. J Mol Biol One of the most widely used bioinformatics applications Alignment quality not as good as Smith-Waterman But much faster, supported at NCBI with big computer cluster For tutorials or information: nformation3.html
51 BLAST Algorithm Steps Query and DB sequences are optionally filtered to remove low-complexity regions E.g. ACACACACA, TTTTTTTTT
52 BLAST Algorithm Steps Query and DB sequences are optionally filtered to remove low-complexity regions Break DB sequences into k-mer words and hash their locations to speed later searches k is usually 11 for DNA/RNA and 3 for protein LPPQGLL LPP PPQ PQG QGL GLL
53 BLAST Algorithm Steps Query and DB sequences are optionally filtered to remove low-complexity regions Break DB sequences into k-mer words and hash their locations to speed later searches Each k-mer in query find possible k-mers that matches well with it well is evaluated by substitution matrices
54 BLAST Algorithm Steps Only words with T cutoff score is kept T is usually 11-13, ~ 50 words make T cutoff Note: this is 50 words at every query position For each DB sequence with a high scoring word, try to extend it in both ends Query: DB seq: LP PQG LL MP PEG LL HSP score = 32 Form HSP (High-scoring Segment Pairs) Use BLOSUM to score the extended alignment No gaps allowed
55 The BLAST Search Algorithm Query: GSVEDTTGSQSLAALLNKCKTPQGQRLVNQWIKQPLMDKNRIEERLNLVEAFVEDAELRQTLQEDL Neighbourhood Words Query Word { PQG 18 PEG 15 PRG 14 PKG 14 PNG 13 PDG 13 PHG 13 PMG 13 PSG 13 PQA 12 PQN 12 Score Threshold (13) Query: 325 SLAALLNKCKTPQGQRLVNQWIKQPLMDKNRIEERLNLVEA 365 +LA++L+ TP G R++ +W+ P+ D + ER + A Sbjct: 290 TLASVLDCTVTPMGSRMLKRWLHMPVRDTRVLLERQQTIGA 330 High-scoring Segment Pair
56 BLAST Algorithm Steps Keep only statistically significant HSPs Based on the scores of aligning 2 random seqs Use Smith-Waterman algorithm to join the HSPs and get optimal alignment Gaps are allowed default (-11, -1)
57 BLAST algorithm summary query subjects (database) seeds (111111) ( ) Scan the index and find all word hits DP extension to recover the high scoring pairs neighborhood words (branch and bound algorithm) Indexing all seeds Extending high scoring pairs Evaluate Significance of HSPs by Karlin-Altschul Statistic: E=KMNexp(-lambda*S)
58 BLAST DB: nr (non-redundant): GenBank, RefSeq, EMBL est: expressed sequences (cdna), redundant Swissprot and pdb: protein databases If query is DNA, but known to be coding (e.g. cdna) Translate cdna into protein Zero gapextension penalty Different BLAST Programs
59 Different BLAST Programs Program blastp blastn blastx tblastn tblastx Description Compares an amino acid query sequence against a protein sequence database. Compares a nucleotide query sequence against a nucleotide sequence database. Compares a nucleotide query sequence translated in all reading frames against a protein sequence database. You could use this option to find potential translation products of an unknown nucleotide sequence. Compares a protein query sequence against a nucleotide sequence database dynamically translated in all reading frames. Compares the six-frame translations of a nucleotide query sequence against the six-frame translations of a nucleotide sequence database. Please note that the tblastx program cannot be used with the nr database on the BLAST Web page because it is too computationally intensive.
60 PSI-BLAST Position Specific Iterative BLAST Align high scoring hits in initial BLAST to construct a profile for the hits Use profile (PSSM) for next iteration BLAST Query Seq2 Seq1 Seq3 Seq4 Find remote homologs or protein families FP sequences can degrade search quickly






61 PSI-BLAST Query GKATFGKLFAAHPEYQQMFRFF Search Initial Matches GKATFGKLFAAHPEYQQMFRFF GKDCLIKFLSAHPQMAAVFGFS GLELWKGILREHPEIKAPFSRV SLHFWKEFLHDHPDLVSLFKRV GFDILISVLDDKPVLDQALAHY PSSM Profile Pos Ala Cys Glu Asp Phe Gly His Search again Refined Matches TSTMYKYMFQTYPEVRSYFNMT GKATFGKLFAAHPEYQQMFRFF SGIAMKRQALVFGAILQEFVAN GKDCLIKFLSAHPQMAAVFGFS WAKASAAWGTAGPEFFMALFDA GLELWKGILREHPEIKAPFSRV SLHFWKEFLHDHPDLVSLFKRV GVALMTTLFADNQETIGYFKRL GFDILISVLDDKPVLDQALAHY VDPHLRMSVHLEPKLWSEFWPI
62 Reciprocal Blast Search for orthologous sequences between two species GeneA in Species1 BLAST Species2 GeneB GeneB in Species2 BLAST Species1 GeneA orthologous GeneA GeneB Also called bi-directional best hit
63 BLAT BLAST-Like Alignment Tool Compare to BLAST, BLAT can align much longer regions (MB) really fast with little resources E.g. can map a sequence to the genome in seconds on one Linux computer Allow big gaps (mrna to genome) Need higher similarity (> 95% for DNA and 80% for proteins) for aligned sequences Basic approach Break long sequence into blocks Index k-mers, typically 8-13 Stitch blocks together for final alignment
64 BLAT: Indexing Genome: cacaattatcacgaccgc 3-mers: cac aat tat cac gac cgc Index: aat 3 gac 12 cac 0,9 tat 6 cgc 15 cdna (mrna -> DNA): aattctcac 3-mers: aat att ttc tct ctc tca cac hits: aat 0,3-3 cac 6,0 6 cac 6,9-3 clump: cacaattatcacgaccgc aattctcac

65 BLAT Example Enter sequence and parameters

66 Get result instantly!! BLAT Example
67 Summary of Fast Search Fast sequence similarity search Break seq, hash DB sub-seq, match sub-seq and extend, use DP for optimal alignment *BLAST, most widely used, many applications with sound statistical foundations *BLAT, align sequence to genome, fast yet need higher similarity
68 BLAST score and significance Report DB sequences above a threshold E value: Number (instead of probability pvalue) of matches expected merely by chance E Kmn e S p(s ³ x)»1- exp[-e -x ] m, n are query and DB length K, are constants Smaller E, more stringent
69 Are these proteins homologs? SEQ 1: RVVNLVPS--FWVLDATYKNYAINYNCDVTYKLY L P W L Y N Y C L SEQ 2: QFFPLMPPAPYWILATDYENLPLVYSCTTFFWLF Probably not (score = 9) SEQ 1: RVVNLVPS--FWVLDATYKNYAINYNCDVTYKLY L P W LDATYKNYA Y C L SEQ 2: QFFPLMPPAPYWILDATYKNYALVYSCTTFFWLF MAYBE (score = 15) SEQ 1: RVVNLVPS--FWVLDATYKNYAINYNCDVTYKLY RVV L PS W LDATYKNYA Y CDVTYKL SEQ 2: RVVPLMPSAPYWILDATYKNYALVYSCDVTYKLF Most likely (score = 24)
70 Significance of scores HPDKKAHSIHAWILSKSKVLEGNTKEVVDNVLKT Homology detection algorithm 45 LENENQGKCTIAEYKYDGKKASVYNSFVSNGVKE Low score = unrelated High score = homologs How high is high enough?
71 Other significance questions Pairwise sequence comparison scores Microarray expression measurements Sequence motif scores Functional assignments of genes Call peaks from ChIP-seq data
72 The null hypothesis We are interested in characterizing the distribution of scores from sequence comparison algorithms. We would like to measure how surprising a given score is, assuming that the two sequences are not related. The assumption is called the null hypothesis. The purpose of most statistical tests is to determine whether the observed results provide a reason to reject the hypothesis that they are merely a product of chance factors.
73 Gaussian vs. Extreme Value Distribution (EVD) Gaussian (Normal) Extreme value
?")
74 Gaussian = 68 in., =3 in. What is the chance of picking a person at least 75 in. tall P(X 75)? ( z score x) x From Table: z=2.33 P=0.01
. (Altschul et al, Nat. Genet.")
75 EVD,,, Each alignment/score that BLAST returns is the very best alignment/score among a large number of alignments/scores for those two sequences (ie and EVD problem). (Altschul et al, Nat. Genet. 1994)
76 Computing a p-value The probability of observing a score >4 is the area under the curve to the right of 4. This probability is called a p-value. p-value = Pr(data null)
77 Scaling the EVD An extreme value distribution derived from, e.g., the Smith-Waterman algorithm will have a characteristic mode μ and scale parameter λ. P x S x 1 exp e These parameters depend upon the size of the query, the size of the target database, the substitution matrix and the gap penalties.
78 An example You run BLAST and get a score of 45. You then run BLAST on a shuffled version of the database, and fit an extreme value distribution to the resulting empirical distribution. The parameters of the EVD are μ = 25 and λ = What is the p-value associated with 45? P P x S x 1 exp e S 45 1 exp e exp e 7 1 exp
79 Summary of statistical significance A distribution plots the frequency of a given type of observation. The area under the distribution is 1. Most statistical tests compare observed data to the expected result according to the null hypothesis. Sequence similarity scores follow an extreme value distribution, which is characterized by a larger tail. The p-value associated with a score is the area under the curve to the right of that score.
80 Applying homology: concept and technology Genome evolution Mammalian genome evolution Human genome variation Cancer genome evolution Gene finding Comparative approaches Ab initio approaches Hidden Markov Model Protein structure Threading Regulatory motif finding Profile comparison Pathway/Network comparison PathBLAST Conservation Ultra conserved elements Human accelerated regions
81 Gene prediction Comparing to a known gene from a different species Using EST evidence (aligning transcript to genome) Predicting from sequence (HHM) Using conservation Signature of coding potential What about RNA gene? Using other genomics signals Specific epigenetic marks of promoters and gene bodies
82 Modeling gene features [human] Exon 1 Exon 2 Exon 3 Intron 1 Intron [mouse] CNS CNS CNS

83 Genscan (Burge and Karlin, 1998) Dramatic improvement over previous methods Generalised HMM Different parameter sets for different GC content regions (intron length distribution and exon stats)

84 Predicting non-coding RNA? From sequence? Not clear which properties can be exploited Sequence features such as promoters are too weak Histone modifications + conservation worked
85
86 So far, only linear sequence comparison C G A A T T C A C A C T T G A A T T C G C C T
87 Expanding the idea of a sequence
88 Central theme of the new algorithm compare profiles T G C A A C G T
 Match = 1 Mismatch =-1")
89 Met14 vs Met2 DotPlot MET14 (1000nt) Match = 1 Mismatch =-1 Gray: 1
90 Met14 vs Met2 MET14 (1000nt) Red: >5
91 Met14 vs Met2 PhyloNet MET14 (1000nt) HSPs: E < 0.1 TTTCACGTGA P=1.75E-5 P=0.002 P=0.003 P=0.03 P=0.02


92 Trey Ideker PathBlast, NetworkBlast

93 Comparative Genomics Functional DNA often evolves slower than neutral DNA. To detect functional elements: align genomes of related species, and find regions of high conservation. The difference between conservation and constraint.


94 Ultra conserved elements ultra conserved e.d 12.5
95

96 HARs: Human accelerated regions 118 bp segment with 18 changes between the human and chimp sequences Expect less than 1
97 Human HAR1F differs from the ancestral RNA stucture
98 Aligning Short Reads




99 Past and Current Sequencing 0 and 1 st generation sequencing Technologies Pre-1992 old fashioned way ABI 373/377 ABI 3700 ABI 3730XL S35 ddntps Gels Manual loading Manual base calling Fluorescent ddntps* Gels Manual loading Automated base calling* Fluorescent ddntps Capillaries* Robotic loading* Automated base calling Breaks down frequently Fluorescent ddntps Capillaries Robotic loading Automated base calling Reliable*









100 Next or 2 nd -generation sequencing Next generation sequencing technology 454/Roche GS-20/FLX (Oct 2005) ABI SOLiD (Oct 2007) Illumina/Solexa 1G Genetic Analyser (Feb 2007)
")

101 8 channels (lanes) Cluster generation Cluster Generation

102 IGA without cover

103 Flowcell imaging Flow cell imaging

104 350 X 350 µm A flow cell A flow cell contains eight lanes Lane 1 Lane 2... Lane 8 Each lane/channel contains three columns of tiles Column 1 Column 2 Column 3 Tile 20K-30K Clusters Each column contains 100 tiles Each tile is imaged four times per cycle one image per base. 345,600 images for a 36-cycle run


105 Data analysis pipeline Data Analysis Pipeline Firecrest Bustard tiff image files (345,600) intensity files Sequence files Additional Data Analysis Alignment to Genome Eland
106 Primary tools and analysis tasks Image processing (unique to each manufacturer) Basecalling (unique to each manufacturer) Align sequence reads to reference genome Assemble contigs and whole genomes using quality scores and/or paired-end information Peak finding for Chip-Seq applications (and statistics to validate, map to regulated genes, etc) SNP calling/genotyping Transcript profiling measure gene expression, identifying alternative splicing, etc.
107 NGS: Sequence alignment Map the large numbers of short reads to a reference genome In a broader sense: Identify similar sequences (DNA, RNA, or protein) in consequence of functional, structural, or evolutionary relationships between the them Applications: Genome assembly, SNP detection, homology search, etc large faster search speed short greater search sensitivity.
108 Mapping Reads Back Hash Table (Lookup table) FAST, but requires perfect matches Array Scanning Can handle mismatches, but not gaps Dynamic Programming (Smith Waterman, Forward, Viterbi) Indels Mathematically optimal solution Slow (most programs use Hash Mapping as a prefilter) Burrows-Wheeler Transform (BW Transform) FAST (memory efficient) But for gaps/mismatches, it lacks sensitivity
109 Many short read aligners
110 Short read mapping Input: A reference genome A collection of many bp tags (reads) User-specified parameters Output: One or more genomic coordinates for each tag In practice, only 70-75% of tags successfully map to the reference genome. Why?
111 Multiple mapping A single tag may occur more than once in the reference genome. The user may choose to ignore tags that appear more than n times. As n gets large, you get more data, but also more noise in the data.
112 Inexact matching? An observed tag may not exactly match any position in the reference genome. Sometimes, the tag almost matches one or more positions. Such mismatches may represent a SNP or a bad readout. The user can specify the maximum number of mismatches, or a phred-style quality score threshold. As the number of allowed mismatches goes up, the number of mapped tags increases, but so does the number of incorrectly mapped tags.
113 Using base qualities to evaluate BETTER! Q: 30 READ: AGGTCCGGGATACCGGGGAC CHR1: CGGTCCGGGATACCGGGGAC BETTER! CHR2: AGGTCCGGGATACCGGGGGT Q: 10+10
114 Hash table (Eland, SOAP) Main idea: preprocess genome to speed up queries Hash every substring of length k K is a tiny constant For each query p, can easily retrieve all suffixes of the genome that start with p1, p2, pk. Easy to implement. Significant speed up in practice. Large memory consumption. Inexact match is difficult. Need multiple hash tables More memory

115 Spaced seed alignment (MAQ) Tags and tag-sized pieces of reference are cut into small seeds. Pairs of spaced seeds are stored in an index. Look up spaced seeds for each tag. For each hit, confirm the remaining positions. Report results to the user.
116 Index the reference genome: Suffix Tree Each suffix corresponds to exactly one path from the root to a leaf Edges spell non-empty strings Construction: linear time and space Check if a string of length m is a substring Each substring is a prefix of a suffix!

117 Burrows-Wheeler (Bowtie, BWA) Store entire reference genome. Align tag base by base from the end. When tag is traversed, all active locations are reported. If no match is found, then back up and try a substitution.
118 Why Burrows-Wheeler? BWT very compact: Approximately ½ byte per base As large as the original text, plus a few extras Can fit onto a standard computer with 2GB of memory Linear-time search algorithm proportional to length of query for exact matches
119 Burrows-Wheeler Transform (BWT) acaacg$ $acaacg aacg$ac acaacg$ acg$aca caacg$a cg$acaa g$acaac BWT gc$aaac Burrows-Wheeler Matrix (BWM)
120 Key observation a 1 c 1 a 2 a 3 c 2 g 1 $ 1 last first (LF) mapping The i-th occurrence of character X in the last column corresponds to the same text character as the i-th occurrence of X in the first column. 1 $acaacg 1 2 aacg$ac 1 1 acaacg$ 1 3 acg$aca 2 1 caacg$a 1 2 cg$acaa 3 1 g$acaac 2
121 Burrows-Wheeler Matrix $acaacg aacg$ac acaacg$ acg$aca caacg$a cg$acaa g$acaac See the suffix array?
122 Burrows-Wheeler Transform Originally designed for data compression for large text Burrows-Wheeler matrix: sort lexicographically all cyclic rotations of S$ BWT(S): the last column of Burrows-Wheeler matrix Compression: runs of repeated characters are easy to compress using move-to-front transform and run-length encoding, etc. BWT(S) is a reversible permutation of S
123 Reverse Burrows-Wheeler Transform BW Matrix Property: Last-First (LF) Mapping The ith occurrence of character X in the last column correspond to the same text character as the ith occurrence of X in the first column


124 qc = the next Searching character to BWT the left in the query Ferragina P, Manzini G: Opportunistic data structures with applications. FOCS. IEEE Computer Society; 2000.
125 Searching BWT BWT(agcagcagact) = tgcc$ggaaaac Search for pattern: gca gca gca gca gca $agcagcagact $agcagcagact $agcagcagact $agcagcagact act$agcagcag act$agcagcag act$agcagcag act$agcagcag agact$agcagc agact$agcagc agact$agcagc agact$agcagc agcagact$agc agcagact$agc agcagact$agc agcagact$agc agcagcagact$ agcagcagact$ agcagcagact$ agcagcagact$ cagact$agcag cagact$agcag cagact$agcag cagact$agcag cagcagact$ag cagcagact$ag cagcagact$ag cagcagact$ag ct$agcagcaga ct$agcagcaga ct$agcagcaga ct$agcagcaga gact$agcagca gact$agcagca gact$agcagca gact$agcagca gcagact$agca gcagact$agca gcagact$agca gcagact$agca gcagcagact$a gcagcagact$a gcagcagact$a gcagcagact$a t$agcagcagac t$agcagcagac t$agcagcagac t$agcagcagac





126 Human genome memory footprint Bowtie Index Suffix Tree Suffix Array Hash Tables 1.3 gigabytes >35 gigabytes >12 gigabytes >12 gigabytes RAM. The Bowtie index can be distributed over the Internet. ficiently on a typical computer with 2 GB 19
DNA and protein databases. EMBL/GenBank/DDBJ database of nucleic acids
 Database searches 1 DNA and protein databases EMBL/GenBank/DDBJ database of nucleic acids 2 DNA and protein databases EMBL/GenBank/DDBJ database of nucleic acids (cntd) 3 DNA and protein databases SWISS-PROT
Database searches 1 DNA and protein databases EMBL/GenBank/DDBJ database of nucleic acids 2 DNA and protein databases EMBL/GenBank/DDBJ database of nucleic acids (cntd) 3 DNA and protein databases SWISS-PROT
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT
 3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Bioinformatics Definitions The use of computational
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Bioinformatics Definitions The use of computational
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment
 Pairwise sequence alignment") Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Introduction to Sequence Alignment. Manpreet S. Katari
 Introduction to Sequence Alignment Manpreet S. Katari 1 Outline 1. Global vs. local approaches to aligning sequences 1. Dot Plots 2. BLAST 1. Dynamic Programming 3. Hash Tables 1. BLAT 4. BWT (Burrow Wheeler
Introduction to Sequence Alignment Manpreet S. Katari 1 Outline 1. Global vs. local approaches to aligning sequences 1. Dot Plots 2. BLAST 1. Dynamic Programming 3. Hash Tables 1. BLAT 4. BWT (Burrow Wheeler
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Bioinformatics and BLAST
 Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
Introduction to Bioinformatics
 Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Basic Local Alignment Search Tool
 Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Sequence Database Search Techniques I: Blast and PatternHunter tools
 Sequence Database Search Techniques I: Blast and PatternHunter tools Zhang Louxin National University of Singapore Outline. Database search 2. BLAST (and filtration technique) 3. PatternHunter (empowered
Sequence Database Search Techniques I: Blast and PatternHunter tools Zhang Louxin National University of Singapore Outline. Database search 2. BLAST (and filtration technique) 3. PatternHunter (empowered
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Biochemistry 324 Bioinformatics. Pairwise sequence alignment
 Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
In-Depth Assessment of Local Sequence Alignment
 2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
Sequence Alignments. Dynamic programming approaches, scoring, and significance. Lucy Skrabanek ICB, WMC January 31, 2013
 Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
BLAST. Varieties of BLAST
 BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
Practical considerations of working with sequencing data
 Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Alignment & BLAST. By: Hadi Mozafari KUMS
 Alignment & BLAST By: Hadi Mozafari KUMS SIMILARITY - ALIGNMENT Comparison of primary DNA or protein sequences to other primary or secondary sequences Expecting that the function of the similar sequence
Alignment & BLAST By: Hadi Mozafari KUMS SIMILARITY - ALIGNMENT Comparison of primary DNA or protein sequences to other primary or secondary sequences Expecting that the function of the similar sequence
Similarity or Identity? When are molecules similar?
 Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I)
 Sequence pairwise alignment (I)") CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
Sequence analysis and Genomics
 Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
BLAST Database Searching. BME 110: CompBio Tools Todd Lowe April 8, 2010
 BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
CONCEPT OF SEQUENCE COMPARISON. Natapol Pornputtapong 18 January 2018
 CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
Alignment principles and homology searching using (PSI-)BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)
BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)") Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Tiffany Samaroo MB&B 452a December 8, Take Home Final. Topic 1
 Tiffany Samaroo MB&B 452a December 8, 2003 Take Home Final Topic 1 Prior to 1970, protein and DNA sequence alignment was limited to visual comparison. This was a very tedious process; even proteins with
Tiffany Samaroo MB&B 452a December 8, 2003 Take Home Final Topic 1 Prior to 1970, protein and DNA sequence alignment was limited to visual comparison. This was a very tedious process; even proteins with
Computational Biology
 Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Whole Genome Alignments and Synteny Maps
 Whole Genome Alignments and Synteny Maps IINTRODUCTION It was not until closely related organism genomes have been sequenced that people start to think about aligning genomes and chromosomes instead of
Whole Genome Alignments and Synteny Maps IINTRODUCTION It was not until closely related organism genomes have been sequenced that people start to think about aligning genomes and chromosomes instead of
Sara C. Madeira. Universidade da Beira Interior. (Thanks to Ana Teresa Freitas, IST for useful resources on this subject)
") Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Grundlagen der Bioinformatik, SS 08, D. Huson, May 2,
 Grundlagen der Bioinformatik, SS 08, D. Huson, May 2, 2008 39 5 Blast This lecture is based on the following, which are all recommended reading: R. Merkl, S. Waack: Bioinformatik Interaktiv. Chapter 11.4-11.7
Grundlagen der Bioinformatik, SS 08, D. Huson, May 2, 2008 39 5 Blast This lecture is based on the following, which are all recommended reading: R. Merkl, S. Waack: Bioinformatik Interaktiv. Chapter 11.4-11.7
Pairwise & Multiple sequence alignments
 Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Sequence Comparison: Local Alignment. Genome 373 Genomic Informatics Elhanan Borenstein
 Sequence Comparison: Local Alignment Genome 373 Genomic Informatics Elhanan Borenstein A quick review: Global Alignment Global Alignment Mission: Fin the best global alignment between two sequences. An
Sequence Comparison: Local Alignment Genome 373 Genomic Informatics Elhanan Borenstein A quick review: Global Alignment Global Alignment Mission: Fin the best global alignment between two sequences. An
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Statistical Machine Learning Methods for Bioinformatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD Department of Computer Science University of Missouri 2008 Free for Academic
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
SEQUENCE ALIGNMENT BACKGROUND: BIOINFORMATICS. Prokaryotes and Eukaryotes. DNA and RNA
 SEQUENCE ALIGNMENT BACKGROUND: BIOINFORMATICS 1 Prokaryotes and Eukaryotes 2 DNA and RNA 3 4 Double helix structure Codons Codons are triplets of bases from the RNA sequence. Each triplet defines an amino-acid.
SEQUENCE ALIGNMENT BACKGROUND: BIOINFORMATICS 1 Prokaryotes and Eukaryotes 2 DNA and RNA 3 4 Double helix structure Codons Codons are triplets of bases from the RNA sequence. Each triplet defines an amino-acid.
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Pairwise sequence alignment
 Department of Evolutionary Biology Example Alignment between very similar human alpha- and beta globins: GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKL G+ +VK+HGKKV A+++++AH+D++ +++++LS+LH KL GNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKL
Department of Evolutionary Biology Example Alignment between very similar human alpha- and beta globins: GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKL G+ +VK+HGKKV A+++++AH+D++ +++++LS+LH KL GNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKL
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Pairwise sequence alignments
 Pairwise sequence alignments Volker Flegel VI, October 2003 Page 1 Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs VI, October
Pairwise sequence alignments Volker Flegel VI, October 2003 Page 1 Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs VI, October
Pairwise sequence alignments. Vassilios Ioannidis (From Volker Flegel )
") Pairwise sequence alignments Vassilios Ioannidis (From Volker Flegel ) Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs Importance
Pairwise sequence alignments Vassilios Ioannidis (From Volker Flegel ) Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs Importance
Single alignment: Substitution Matrix. 16 march 2017
 Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences
 Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
Statistical Machine Learning Methods for Biomedical Informatics II. Hidden Markov Model for Biological Sequences Jianlin Cheng, PhD William and Nancy Thompson Missouri Distinguished Professor Department
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 05: Index-based alignment algorithms Slides adapted from Dr. Shaojie Zhang (University of Central Florida) Real applications of alignment Database search
EECS730: Introduction to Bioinformatics Lecture 05: Index-based alignment algorithms Slides adapted from Dr. Shaojie Zhang (University of Central Florida) Real applications of alignment Database search
Pairwise Sequence Alignment
 Introduction to Bioinformatics Pairwise Sequence Alignment Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Outline Introduction to sequence alignment pair wise sequence alignment The Dot Matrix Scoring
Introduction to Bioinformatics Pairwise Sequence Alignment Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Outline Introduction to sequence alignment pair wise sequence alignment The Dot Matrix Scoring
Pairwise Alignment. Guan-Shieng Huang. Dept. of CSIE, NCNU. Pairwise Alignment p.1/55
 Pairwise Alignment Guan-Shieng Huang shieng@ncnu.edu.tw Dept. of CSIE, NCNU Pairwise Alignment p.1/55 Approach 1. Problem definition 2. Computational method (algorithms) 3. Complexity and performance Pairwise
Pairwise Alignment Guan-Shieng Huang shieng@ncnu.edu.tw Dept. of CSIE, NCNU Pairwise Alignment p.1/55 Approach 1. Problem definition 2. Computational method (algorithms) 3. Complexity and performance Pairwise
1.5 Sequence alignment
 1.5 Sequence alignment The dramatic increase in the number of sequenced genomes and proteomes has lead to development of various bioinformatic methods and algorithms for extracting information (data mining)
1.5 Sequence alignment The dramatic increase in the number of sequenced genomes and proteomes has lead to development of various bioinformatic methods and algorithms for extracting information (data mining)
Heuristic Alignment and Searching
 3/28/2012 Types of alignments Global Alignment Each letter of each sequence is aligned to a letter or a gap (e.g., Needleman-Wunsch). Local Alignment An optimal pair of subsequences is taken from the two
3/28/2012 Types of alignments Global Alignment Each letter of each sequence is aligned to a letter or a gap (e.g., Needleman-Wunsch). Local Alignment An optimal pair of subsequences is taken from the two
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Motivating the need for optimal sequence alignments...
 1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
Practical Bioinformatics
 5/2/2017 Dictionaries d i c t i o n a r y = { A : T, T : A, G : C, C : G } d i c t i o n a r y [ G ] d i c t i o n a r y [ N ] = N d i c t i o n a r y. h a s k e y ( C ) Dictionaries g e n e t i c C o
5/2/2017 Dictionaries d i c t i o n a r y = { A : T, T : A, G : C, C : G } d i c t i o n a r y [ G ] d i c t i o n a r y [ N ] = N d i c t i o n a r y. h a s k e y ( C ) Dictionaries g e n e t i c C o
Collected Works of Charles Dickens
 Collected Works of Charles Dickens A Random Dickens Quote If there were no bad people, there would be no good lawyers. Original Sentence It was a dark and stormy night; the night was dark except at sunny
Collected Works of Charles Dickens A Random Dickens Quote If there were no bad people, there would be no good lawyers. Original Sentence It was a dark and stormy night; the night was dark except at sunny
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Sequences, Structures, and Gene Regulatory Networks
 Sequences, Structures, and Gene Regulatory Networks Learning Outcomes After this class, you will Understand gene expression and protein structure in more detail Appreciate why biologists like to align
Sequences, Structures, and Gene Regulatory Networks Learning Outcomes After this class, you will Understand gene expression and protein structure in more detail Appreciate why biologists like to align
Sequence Analysis 17: lecture 5. Substitution matrices Multiple sequence alignment
 Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Introduction to sequence alignment. Local alignment the Smith-Waterman algorithm
 Lecture 2, 12/3/2003: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties Local alignment the Smith-Waterman algorithm 1 Computational
Lecture 2, 12/3/2003: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties Local alignment the Smith-Waterman algorithm 1 Computational
InDel 3-5. InDel 8-9. InDel 3-5. InDel 8-9. InDel InDel 8-9
 Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
O 3 O 4 O 5. q 3. q 4. Transition
 Hidden Markov Models Hidden Markov models (HMM) were developed in the early part of the 1970 s and at that time mostly applied in the area of computerized speech recognition. They are first described in
Hidden Markov Models Hidden Markov models (HMM) were developed in the early part of the 1970 s and at that time mostly applied in the area of computerized speech recognition. They are first described in
Comparing whole genomes
 BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
Alignment Algorithms. Alignment Algorithms
 Midterm Results Big improvement over scores from the previous two years. Since this class grade is based on the previous years curve, that means this class will get higher grades than the previous years.
Midterm Results Big improvement over scores from the previous two years. Since this class grade is based on the previous years curve, that means this class will get higher grades than the previous years.
Administration. ndrew Torda April /04/2008 [ 1 ]
![Administration. ndrew Torda April /04/2008 [ 1 ]](/thumbs/73/68891262.jpg "Administration. ndrew Torda April /04/2008 [ 1 ]") ndrew Torda April 2008 Administration 22/04/2008 [ 1 ] Sprache? zu verhandeln (Englisch, Hochdeutsch, Bayerisch) Selection of topics Proteins / DNA / RNA Two halves to course week 1-7 Prof Torda (larger
ndrew Torda April 2008 Administration 22/04/2008 [ 1 ] Sprache? zu verhandeln (Englisch, Hochdeutsch, Bayerisch) Selection of topics Proteins / DNA / RNA Two halves to course week 1-7 Prof Torda (larger
Protein Bioinformatics. Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet sandberg.cmb.ki.
 Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
Protein function prediction based on sequence analysis
 Performing sequence searches Post-Blast analysis, Using profiles and pattern-matching Protein function prediction based on sequence analysis Slides from a lecture on MOL204 - Applied Bioinformatics 18-Oct-2005
Performing sequence searches Post-Blast analysis, Using profiles and pattern-matching Protein function prediction based on sequence analysis Slides from a lecture on MOL204 - Applied Bioinformatics 18-Oct-2005
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences
 Analysis of Biological Sequences") First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
HMMs and biological sequence analysis
 HMMs and biological sequence analysis Hidden Markov Model A Markov chain is a sequence of random variables X 1, X 2, X 3,... That has the property that the value of the current state depends only on the
HMMs and biological sequence analysis Hidden Markov Model A Markov chain is a sequence of random variables X 1, X 2, X 3,... That has the property that the value of the current state depends only on the
SUPPLEMENTARY INFORMATION
 Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Local Alignment Statistics
 Local Alignment Statistics Stephen Altschul National Center for Biotechnology Information National Library of Medicine National Institutes of Health Bethesda, MD Central Issues in Biological Sequence Comparison
Local Alignment Statistics Stephen Altschul National Center for Biotechnology Information National Library of Medicine National Institutes of Health Bethesda, MD Central Issues in Biological Sequence Comparison
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment
 Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, Global and local alignment of two sequences using dynamic programming
 20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, 2008 4 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance 4. Global and local alignment
20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, 2008 4 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance 4. Global and local alignment
Bioinformatics. Part 8. Sequence Analysis An introduction. Mahdi Vasighi
 Bioinformatics Sequence Analysis An introduction Part 8 Mahdi Vasighi Sequence analysis Some of the earliest problems in genomics concerned how to measure similarity of DNA and protein sequences, either
Bioinformatics Sequence Analysis An introduction Part 8 Mahdi Vasighi Sequence analysis Some of the earliest problems in genomics concerned how to measure similarity of DNA and protein sequences, either
Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm. Alignment scoring schemes and theory: substitution matrices and gap models
 Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm Alignment scoring schemes and theory: substitution matrices and gap models 1 Local sequence alignments Local sequence alignments are necessary
Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm Alignment scoring schemes and theory: substitution matrices and gap models 1 Local sequence alignments Local sequence alignments are necessary
Linear-Space Alignment
 Linear-Space Alignment Subsequences and Substrings Definition A string x is a substring of a string x, if x = ux v for some prefix string u and suffix string v (similarly, x = x i x j, for some 1 i j x
Linear-Space Alignment Subsequences and Substrings Definition A string x is a substring of a string x, if x = ux v for some prefix string u and suffix string v (similarly, x = x i x j, for some 1 i j x
Lecture 5,6 Local sequence alignment
 Lecture 5,6 Local sequence alignment Chapter 6 in Jones and Pevzner Fall 2018 September 4,6, 2018 Evolution as a tool for biological insight Nothing in biology makes sense except in the light of evolution
Lecture 5,6 Local sequence alignment Chapter 6 in Jones and Pevzner Fall 2018 September 4,6, 2018 Evolution as a tool for biological insight Nothing in biology makes sense except in the light of evolution
GEP Annotation Report
 GEP Annotation Report Note: For each gene described in this annotation report, you should also prepare the corresponding GFF, transcript and peptide sequence files as part of your submission. Student name:
GEP Annotation Report Note: For each gene described in this annotation report, you should also prepare the corresponding GFF, transcript and peptide sequence files as part of your submission. Student name:
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5
 Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
MATHEMATICAL MODELS - Vol. III - Mathematical Modeling and the Human Genome - Hilary S. Booth MATHEMATICAL MODELING AND THE HUMAN GENOME
 MATHEMATICAL MODELING AND THE HUMAN GENOME Hilary S. Booth Australian National University, Australia Keywords: Human genome, DNA, bioinformatics, sequence analysis, evolution. Contents 1. Introduction:
MATHEMATICAL MODELING AND THE HUMAN GENOME Hilary S. Booth Australian National University, Australia Keywords: Human genome, DNA, bioinformatics, sequence analysis, evolution. Contents 1. Introduction:
Phylogenetic inference
 Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Lecture 2: Pairwise Alignment. CG Ron Shamir
 Lecture 2: Pairwise Alignment 1 Main source 2 Why compare sequences? Human hexosaminidase A vs Mouse hexosaminidase A 3 www.mathworks.com/.../jan04/bio_genome.html Sequence Alignment עימוד רצפים The problem:
Lecture 2: Pairwise Alignment 1 Main source 2 Why compare sequences? Human hexosaminidase A vs Mouse hexosaminidase A 3 www.mathworks.com/.../jan04/bio_genome.html Sequence Alignment עימוד רצפים The problem:
Sequence alignment methods. Pairwise alignment. The universe of biological sequence analysis
 he universe of biological sequence analysis Word/pattern recognition- Identification of restriction enzyme cleavage sites Sequence alignment methods PstI he universe of biological sequence analysis - prediction
he universe of biological sequence analysis Word/pattern recognition- Identification of restriction enzyme cleavage sites Sequence alignment methods PstI he universe of biological sequence analysis - prediction
C E N T R. Introduction to bioinformatics 2007 E B I O I N F O R M A T I C S V U F O R I N T. Lecture 5 G R A T I V. Pair-wise Sequence Alignment
 C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 5 Pair-wise Sequence Alignment Bioinformatics Nothing in Biology makes sense except in
C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 5 Pair-wise Sequence Alignment Bioinformatics Nothing in Biology makes sense except in
EECS730: Introduction to Bioinformatics
 EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
EECS730: Introduction to Bioinformatics Lecture 07: profile Hidden Markov Model http://bibiserv.techfak.uni-bielefeld.de/sadr2/databasesearch/hmmer/profilehmm.gif Slides adapted from Dr. Shaojie Zhang
Bioinformatics Exercises
 Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
Mathangi Thiagarajan Rice Genome Annotation Workshop May 23rd, 2007
 -2 Transcript Alignment Assembly and Automated Gene Structure Improvements Using PASA-2 Mathangi Thiagarajan mathangi@jcvi.org Rice Genome Annotation Workshop May 23rd, 2007 About PASA PASA is an open
-2 Transcript Alignment Assembly and Automated Gene Structure Improvements Using PASA-2 Mathangi Thiagarajan mathangi@jcvi.org Rice Genome Annotation Workshop May 23rd, 2007 About PASA PASA is an open
Sequence Alignment (chapter 6)
") Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
Comparative Gene Finding. BMI/CS 776 Spring 2015 Colin Dewey
 Comparative Gene Finding BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2015 Colin Dewey cdewey@biostat.wisc.edu Goals for Lecture the key concepts to understand are the following: using related genomes
Comparative Gene Finding BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2015 Colin Dewey cdewey@biostat.wisc.edu Goals for Lecture the key concepts to understand are the following: using related genomes
Genome Annotation. Qi Sun Bioinformatics Facility Cornell University
 Genome Annotation Qi Sun Bioinformatics Facility Cornell University Some basic bioinformatics tools BLAST PSI-BLAST - Position-Specific Scoring Matrix HMM - Hidden Markov Model NCBI BLAST How does BLAST
Genome Annotation Qi Sun Bioinformatics Facility Cornell University Some basic bioinformatics tools BLAST PSI-BLAST - Position-Specific Scoring Matrix HMM - Hidden Markov Model NCBI BLAST How does BLAST
Study and Implementation of Various Techniques Involved in DNA and Protein Sequence Analysis
 Study and Implementation of Various Techniques Involved in DNA and Protein Sequence Analysis Kumud Joseph Kujur, Sumit Pal Singh, O.P. Vyas, Ruchir Bhatia, Varun Singh* Indian Institute of Information
Study and Implementation of Various Techniques Involved in DNA and Protein Sequence Analysis Kumud Joseph Kujur, Sumit Pal Singh, O.P. Vyas, Ruchir Bhatia, Varun Singh* Indian Institute of Information
Sequence Comparison. mouse human
 Sequence Comparison Sequence Comparison mouse human Why Compare Sequences? The first fact of biological sequence analysis In biomolecular sequences (DNA, RNA, or amino acid sequences), high sequence similarity
Sequence Comparison Sequence Comparison mouse human Why Compare Sequences? The first fact of biological sequence analysis In biomolecular sequences (DNA, RNA, or amino acid sequences), high sequence similarity
Bio 1B Lecture Outline (please print and bring along) Fall, 2007
 Fall, 2007") Bio 1B Lecture Outline (please print and bring along) Fall, 2007 B.D. Mishler, Dept. of Integrative Biology 2-6810, bmishler@berkeley.edu Evolution lecture #5 -- Molecular genetics and molecular evolution
Bio 1B Lecture Outline (please print and bring along) Fall, 2007 B.D. Mishler, Dept. of Integrative Biology 2-6810, bmishler@berkeley.edu Evolution lecture #5 -- Molecular genetics and molecular evolution
Biologically significant sequence alignments using Boltzmann probabilities
 Biologically significant sequence alignments using Boltzmann probabilities P. Clote Department of Biology, Boston College Gasson Hall 416, Chestnut Hill MA 02467 clote@bc.edu May 7, 2003 Abstract In this
Biologically significant sequence alignments using Boltzmann probabilities P. Clote Department of Biology, Boston College Gasson Hall 416, Chestnut Hill MA 02467 clote@bc.edu May 7, 2003 Abstract In this
An Introduction to Bioinformatics Algorithms Hidden Markov Models
 Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Hidden Markov Models Outline 1. CG-Islands 2. The Fair Bet Casino 3. Hidden Markov Model 4. Decoding Algorithm 5. Forward-Backward Algorithm 6. Profile HMMs 7. HMM Parameter Estimation 8. Viterbi Training
Advanced topics in bioinformatics
 Feinberg Graduate School of the Weizmann Institute of Science Advanced topics in bioinformatics Shmuel Pietrokovski & Eitan Rubin Spring 2003 Course WWW site: http://bioinformatics.weizmann.ac.il/courses/atib
Feinberg Graduate School of the Weizmann Institute of Science Advanced topics in bioinformatics Shmuel Pietrokovski & Eitan Rubin Spring 2003 Course WWW site: http://bioinformatics.weizmann.ac.il/courses/atib
Genomics and bioinformatics summary. Finding genes -- computer searches
 Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Alignment Strategies for Large Scale Genome Alignments
 Alignment Strategies for Large Scale Genome Alignments CSHL Computational Genomics 9 November 2003 Algorithms for Biological Sequence Comparison algorithm value scoring gap time calculated matrix penalty
Alignment Strategies for Large Scale Genome Alignments CSHL Computational Genomics 9 November 2003 Algorithms for Biological Sequence Comparison algorithm value scoring gap time calculated matrix penalty
CSE : Computational Issues in Molecular Biology. Lecture 6. Spring 2004
 CSE 397-497: Computational Issues in Molecular Biology Lecture 6 Spring 2004-1 - Topics for today Based on premise that algorithms we've studied are too slow: Faster method for global comparison when sequences
CSE 397-497: Computational Issues in Molecular Biology Lecture 6 Spring 2004-1 - Topics for today Based on premise that algorithms we've studied are too slow: Faster method for global comparison when sequences
Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Sepp Hochreiter
 Sepp Hochreiter") Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Institute of Bioinformatics Johannes Kepler University, Linz, Austria Sequence Alignment 2. Sequence Alignment Sequence Alignment 2.1
Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Institute of Bioinformatics Johannes Kepler University, Linz, Austria Sequence Alignment 2. Sequence Alignment Sequence Alignment 2.1