Method Article. Preprints ( NOT PEER-REVIEWED Posted: 17 December 2018
|
|
|
- Sherilyn West
- 5 years ago
- Views:
Transcription
1 Method Article *Title: HoSeIn: a workflow for integrating various homology search results from a high-throughput sequence dataset *Authors: Christina B. McCarthy1,2*, Gastón Rozadilla1 and Jorgelina Moreiras Clemente1 *Affiliations: 1 Centro Regional de Estudios Genómicos, Facultad de Ciencias Exactas, Universidad Nacional de La Plata, La Plata, Argentina. 2 Departamento de Informática y Tecnología, Universidad Nacional del Noroeste de la Provincia de Buenos Aires, Pergamino, Buenos Aires, Argentina. *Contact mccarthychristina@gmail.com (corresponding author*) *Keywords: Metagenomics; Metatranscriptomics; Environmental sample; Homology searches; Taxonomic profile; Functional profile ABSTRACT Data generated by metagenomic and metatranscriptomic experiments is both enormous and inherently noisy [1]. When using taxonomy-dependent alignment-based methods to classify and label reads, such as MEGAN [2], the first step consists in performing homology searches against sequence databases. To obtain the most information from the samples, nucleotide sequences are usually compared to various databases (i.e., nucleotide and protein) using local sequence aligners such as BLASTN and BLASTX [3]. Nevertheless, the analysis and integration of these results can be problematic because the outputs from these searches usually show differences, which can be notorious when working with RNA-seq (Personal observation; Graphical abstract). These inconsistencies led us to develop the HoSeIn workflow to determine the unequivocal taxonomic and functional profile of environmental samples, based on the assumption that the sequences that correspond to a certain taxon are composed of (Graphical abstract): 1) sequences that were assigned to the same taxon by both homology searches, plus 2) sequences that were assigned to that taxon by one of the homology searches but returned no hits in the other one, and vice versa. SPECIFICATIONS TABLE Subject Area Agricultural and Biological Sciences More specific subject area: Metagenomics, Metatranscriptomics Method name: HoSeIn: Homology Search Integration Select one of the following subject areas: Agricultural and Biological Sciences Biochemistry, Genetics and Molecular Biology Chemical Engineering Chemistry Computer Science Earth and Planetary Sciences Energy Engineering Environmental Science Immunology and Microbiology Materials Science Mathematics Medicine and Dentistry Neuroscience Pharmacology, Toxicology and Pharmaceutical Science Physics and Astronomy Psychology Social Sciences Veterinary Science and Veterinary Medicine Describe narrower subject area Please specify a name of the method that you customized. The method name should be a word or short phrase to describe the methods used in your paper Name and reference of original method If applicable, include full bibliographic details of the main reference(s) describing the original method from which the new method was derived by the author(s). Distributed under a Creative Commons CC BY license.
2 Resource availability ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/latest/ If applicable, include links to resources necessary to reproduce the method (e.g. data, software, hardware, reagent) *Method details Note: This protocol describes the global procedure for analysing high-throughput metatranscriptomic and metagenomic sequences from an environmental sample, and focuses in detail on how to define its unequivocal taxonomic and functional profile. It does not include a detailed description of the pre-processing of high-throughput sequences obtained from an environmental sample (for this, see [4][5]), nor on how to perform the homology searches (for this, see [5]), as this will vary according to the Next Generation Sequencing (NGS) method used; neither does it include a detailed description of how to use MEGAN (for this, see [2,6] and the MEGAN user manual). Overview of the HoSeIn workflow: Trimmed sequences from an environmental sample must first be compared to the chosen nucleotide (usually nt; non-redundant) and protein (usually nr; non-redundant) databases using BLASTN [3] and BLASTX [7], respectively. BLASTX [7] searches protein databases using a translated nucleotide sequence. MEGAN (MEtaGenome ANalyzer) [2,6] is a stand-alone interactive tool that uses homology search results as input, to analyse the taxonomic content of metagenomic and metatranscriptomic datasets, as well as their functional profile using SEED [8], COG [9] and KEGG [10]. The graphical abstract shows an overview of the rationale supporting the sequence analysis workflow that we developed, and summarises the different steps that were followed: 1) Sequences were submitted to homology searches against nucleotide and protein databases using BLASTN (BN) and BLASTX (BX), respectively. These homology search results were then analysed with MEGAN and, after performing a global comparison (using one of MEGAN s features), it was clear that they showed differences and inconsistencies (Graphical abstract). For this reason, a workflow was developed to intersect the information from both analyses in order to define the unequivocal taxonomic and functional profile of the sample, based on the assumption that the sequences that correspond to a certain taxon are composed of (Graphical abstract): a) sequences that were assigned to the same taxon by both homology searches, plus b) sequences that were assigned to that taxon by one of the homology searches but returned no hits in the other one, and vice versa. 2) In order to be able to access, visualise, compare and combine the information from both homology searches easily, and so identify and extract the total sequences that corresponded to each taxon, the database (DB) browser for SQLite was used ( This is an open source tool for users and developers wanting to create databases, search, and edit data, that uses a spreadsheet-like interface. For this, the taxonomic and functional information from both homology searches for all taxa was extracted individually from MEGAN (using one of its features), and then combined in the DB browser for SQLite. 3) After creating the database in the SQLite DB browser, the criss-crossing mentioned in 1) was performed in the browser, which enabled us to elucidate the inconsistencies observed in the homology search results, based on the aforementioned assumptions. In this way, the results from each homology search were visible for each sequence in a table, as well as their final taxonomic and functional assignment resulting from the data intersection. Thus was the unequivocal taxonomic and functional profile of the sample determined. HoSeIn workflow tutorial: What follows is a detailed tutorial in which we exemplify our HoSeIn workflow. To be able to follow the step-by-step tutorial, we provide sample data in the form of the "RMA" files generated by MEGAN6 after it processes the text files from the homology searches (Supplementary material: blastn_nt16slep_total-contigs.rma6 and blastx_nr_total-contigs.rma6). The sequences that were used in the homology searches originated from total RNA extracted from an insect pest gut, which was pyrosequenced [11] and later assembled into contigs. A few remarks follow to explain certain features that are distinctive of this particular sample: The purpose of the mentioned study was to integrate gene expression data from Spodoptera frugiperda guts and their associated metatranscriptomes. For this, total RNA was extracted from fifth instar larval guts and submitted to a one-step reverse transcription and PCR sequence-independent amplification procedure, and then pyrosequenced [11]; the high-throughput reads were later assembled into contigs. As we were interested in identifying and differentiating the host (S. frugiperda) gut transcriptome as well as its associated metatranscriptome, the following NCBI databases were downloaded locally for the homology searches (ftp://ftp.ncbi.nlm.nih.gov/blast/db/): i) Nucleotide: - Non-redundant nucleotide sequence (nt), - 16S microbial (16S), - Lepidopteran whole genome shotgun (Lep) projects completed at the time of the analysis. Sequences from nt, 16S, and Lep, were then combined in a single database (DB:nt16SLep) using the appropriate BLAST+ applications (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/latest/).
.")
.")
3 ii) Protein: - non-redundant protein sequence (nr). Total contigs were then compared to the combined nucleotide database (db: nt16slep) using BLASTN, and to the protein database (nr) using BLASTX. The Lep sequences of the combined nucleotide database simplified the identification of sequences that corresponded to the host (which represented the vast majority). The nt and 16S databases in the combined nucleotide database (db: nt16slep) enabled the identification of the associated metatranscriptome. In this tutorial we only show how we determined the taxonomical identity of these contigs (i.e., we do not include the identification of the functional profile as well). As mentioned previously, we provide sample data in the form of "RMA" files (Supplementary material: blastn_nt16slep_total-contigs.rma6 and blastx_nr_total-contigs.rma6) to be able to follow the step-by-step tutorial from the point in which the taxonomical information is extracted from MEGAN. I) How to download MEGAN: Summary: MEGAN must be downloaded and installed locally to be able to process the text files from the homology searches and then extract the taxonomical and functional information. Step by step tutorial: MEGAN installation: Download the MEGAN6 version that matches your operating system, and the mapping files (Protein accession to NCBI-taxonomy mapping file, and Nucleotide accession to NCBI-taxonomy mapping file):
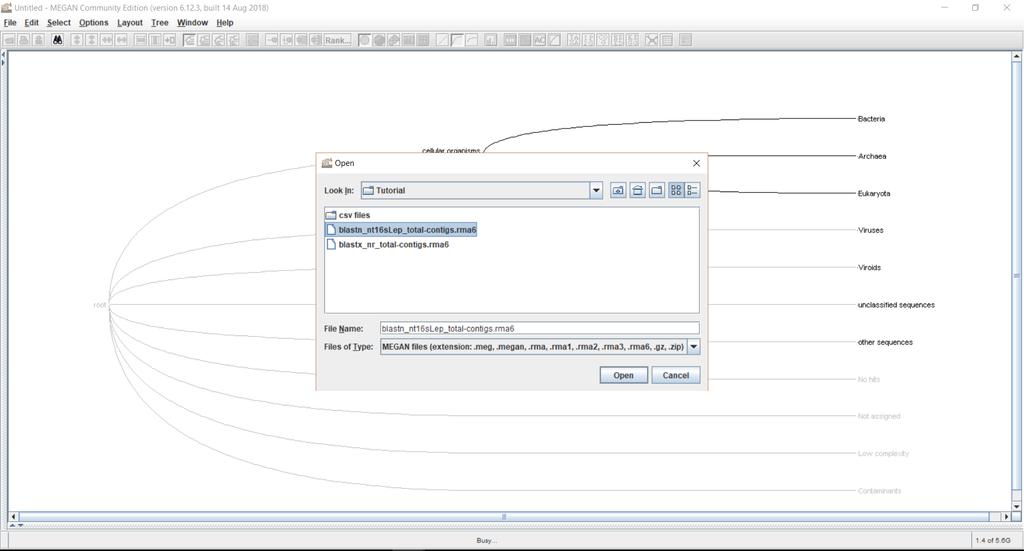
4 Run the installer. This is the MEGAN6 main window: Open the provided RMA files (blastn_nt16slep_total-contigs.rma6 and blastx_nr_total-contigs.rma6):
5
 format. In each of these files there are two columns, one with the contig name (e.g., Contig479) and another with the corresponding assigned taxonomic level (Domain, Phylum, etc.")
6 II) How to extract the information from MEGAN: Summary: To extract the taxonomic information from MEGAN, the taxonomic tree must be progressively expanded from Domain to species, selecting all the leaves, and extracting the text files in csv (comma-separated values) format. In each of these files there are two columns, one with the contig name (e.g., Contig479) and another with the corresponding assigned taxonomic level (Domain, Phylum, etc.). In this way, a series of files are obtained from both the BLASTN and BLASTX homology searches (blastn_domain, blastn_phylum, blastx_domain, blastx_phylum, etc.). Step by step tutorial: Choose the taxonomic level that you wish to extract (from Domain to Species): Tree > Rank Here we show the tree collapsed to Domain level:
7 To select the leaves: Select > All Leaves Without deselecting the leaves, export the file to csv format: File > Export > CSV Format
.")
8 Choose what data you want to export and in what way it will be tabbed in the csv file (choose Summarised so it exports the sequences contained in the chosen taxonomic level as well as all the lower levels). In this way a csv text file is obtained (which can be viewed in a basic word processor such as WordPad):
9 Repeat this procedure for each taxonomic level, and for the other homology search. In this way, 14 total files are obtained (7 files for each homology search), each one corresponding to a particular taxonomic level: III) How to combine the information extracted from MEGAN to create a database in the DB browser for SQLite: Below is a brief explanation of the DB browser for SQLite commands that were used to create the database: - Naming tables and columns: the table name is separated from the column name by a period, e.g. taxonomy.blastn-domain indicates the blastn-domain column from the taxonomy table. - SELECT table1.column1, table2.column2, etc.: indicates what columns we want to visualise, using the naming system explained in the previous paragraph.
. - WHERE table1.")
10 - FROM table1: indicates what table to retrieve the columns from. - LEFT JOIN table2 ON table2.contig = table1.contig: to join columns from various tables following a certain criterion (in this example, the contig number). - WHERE table1.column1 LIKE Arthropoda : to filter the table following a certain criterion; in this example, to only visualise those contigs that were assigned to Arthropoda. - UPDATE column1 SET value1 WHERE condition1: to update the information in column1 with value1 when a certain condition is fulfilled. Summary: The next step consisted in combining all the files extracted from MEGAN to create a single table which included both taxonomic assignments for each contig (the one from BLASTN and the one from BLASTX). To do this, each csv file was imported individually to the DB browser for SQLite, using the following commands: SELECT blastn_domain.contig,blastx_domain.blastx_domain,blastx_phylum.blastx_phylum,blastx_class.blastx_class,blastx_order.blastx_or der,blastx_family.blastx_family,blastx_genus.blastx_genus,blastx_species.blastx_species,blastn_domain.blastn_domain,blastn_ph ylum.blastn_phylum,blastn_class.blastn_class,blastn_order.blastn_order,blastn_family.blastn_family,blastn_genus.blastn_genus,bl astn_species.blastn_species FROM blastn_domain LEFT JOIN blastx_domain ON blastx_domain.contig = blastn_domain.contig LEFT JOIN blastx_phylum ON blastx_phylum.contig = blastn_domain.contig LEFT JOIN blastx_class ON blastx_class.contig = blastn_domain.contig LEFT JOIN blastx_order ON blastx_order.contig = blastn_domain.contig LEFT JOIN blastx_family ON blastx_family.contig = blastn_domain.contig LEFT JOIN blastx_genus ON blastx_genus.contig = blastn_domain.contig LEFT JOIN blastx_species ON blastx_species.contig = blastn_domain.contig LEFT JOIN blastn_phylum ON blastn_phylum.contig = blastn_domain.contig LEFT JOIN blastn_class ON blastn_class.contig = blastn_domain.contig LEFT JOIN blastn_order ON blastn_order.contig = blastn_domain.contig LEFT JOIN blastn_family ON blastn_family.contig = blastn_domain.contig LEFT JOIN blastn_genus ON blastn_genus.contig = blastn_domain.contig LEFT JOIN blastn_species ON blastn_species.contig = blastn_domain.contig To simplify the naming of tables and columns, this result was exported in csv format and imported again to the DB browser for SQLite as the Taxonomy table. Various empty columns were added to this table (named final_domain, final_phylum, etc.) to be completed with the final result of the criss-crossing of the assignments, and another column state to indicate if the contig was assigned or not (to avoid multiple assignments). Step by step tutorial: DB Browser for SQLite installation: DB Browser for SQLite main window:


11 Create a new database clicking on New Database and choosing where to save it. Import csv files that were exported from MEGAN6: File > Import > Table from CSV file Choose a representative name for the table (in this example, blastn_domain), and indicate field separator (in this case, Tab):
.")

12 To simplify the interpretation of the commands and avoid mistakes, we recommend renaming the table columns with representative names (in this example, contig and blastn_domain). For this, in the main window select the table that will be modified and click Modify table. A window will open where both fields appear. Double click on each to rename: Repeat this procedure for each of the 14 csv files:


13 In the Execute SQL leaf, paste the following commands and execute them with Play : SELECT blastn_domain.contig,blastx_domain.blastx_domain,blastx_phylum.blastx_phylum,blastx_class.blastx_class,blastx_order.blastx_or der,blastx_family.blastx_family,blastx_genus.blastx_genus,blastx_species.blastx_species,blastn_domain.blastn_domain,blastn_ph ylum.blastn_phylum,blastn_class.blastn_class,blastn_order.blastn_order,blastn_family.blastn_family,blastn_genus.blastn_genus,bl astn_species.blastn_species FROM blastn_domain LEFT JOIN blastx_domain ON blastx_domain.contig = blastn_domain.contig LEFT JOIN blastx_phylum ON blastx_phylum.contig = blastn_domain.contig LEFT JOIN blastx_class ON blastx_class.contig = blastn_domain.contig LEFT JOIN blastx_order ON blastx_order.contig = blastn_domain.contig LEFT JOIN blastx_family ON blastx_family.contig = blastn_domain.contig LEFT JOIN blastx_genus ON blastx_genus.contig = blastn_domain.contig LEFT JOIN blastx_species ON blastx_species.contig = blastn_domain.contig LEFT JOIN blastn_phylum ON blastn_phylum.contig = blastn_domain.contig LEFT JOIN blastn_class ON blastn_class.contig = blastn_domain.contig LEFT JOIN blastn_order ON blastn_order.contig = blastn_domain.contig LEFT JOIN blastn_family ON blastn_family.contig = blastn_domain.contig LEFT JOIN blastn_genus ON blastn_genus.contig = blastn_domain.contig LEFT JOIN blastn_species ON blastn_species.contig = blastn_domain.contig


14 Once it has been executed, the result will be shown in the bottom part of the window. Now this result can be saved in the database as View, or it can be exported in csv format to be analysed with other programs. To do this, we click on the save button and choose the desired option: In this example, Export to CSV to obtain the complete table: Having obtained the taxonomy.csv file, we now import it to the database to be able to perform the criss-crossing. Having selected the taxonomy table in the main window, we now choose Modify Table as indicated previously:
, to be completed with the crisscrossing result, and another column state was added to indicate if the contig was assigned or not (to")
 Intersection of taxonomic data: Summary: The taxonomy database created in")
15 In this example, 7 columns were added to the taxonomy table (final_domain, final_phylum, etc.), to be completed with the crisscrossing result, and another column state was added to indicate if the contig was assigned or not (to avoid multiple assignments). To add more columns to the table, click on Add field : IV) Intersection of taxonomic data: Summary: The taxonomy database created in the DB browser for SQLite was then used to elucidate the inconsistencies observed in the homology search results, based on the aforementioned assumptions.

16 Step by step tutorial: a) Contigs that showed hits against lepidopterans in the nt16slep homology search, were directly annotated with the taxonomic assignment of that homology search using the following commands: SET final_domain = blastn_domain, final_phylum = blastn_phylum, final_class = blastn_class, final_order = blastn_order, final_family = blastn_family, final_genus = blastn_genus, final_species = blastn_species, state = "assigned" WHERE blastn_phylum LIKE "Arthropoda" b) Contigs that were assigned to the same taxon by both homology searches, were annotated with the following commands: SET final_domain = blastx_domain WHERE state isnull AND blastx_domain=blastn_domain; SET final_phylum = blastx_phylum WHERE state isnull AND blastx_phylum=blastn_phylum; SET final_class = blastx_class WHERE state isnull AND blastx_class=blastn_class; SET final_order = blastx_order WHERE state isnull AND blastx_order=blastn_order; SET final_family = blastx_family WHERE state isnull AND blastx_family=blastn_family; SET final_genus = blastx_genus WHERE state isnull AND blastx_genus=blastn_genus; SET final_species = blastx_species WHERE state isnull AND blastx_species=blastn_species; SET state = "assigned" WHERE state isnull AND final_domain not LIKE "";
17 c) Contigs that showed hits in only one of the homology searches (and no hits in the other one), were annotated with the following commands: SET final_domain = blastx_domain WHERE state isnull AND blastn_domain LIKE ""; SET final_phylum = blastx_phylum WHERE state isnull AND blastn_phylum LIKE ""; SET final_class = blastx_class WHERE state isnull AND blastn_class LIKE ""; SET final_order = blastx_order WHERE state isnull AND blastn_order LIKE ""; SET final_family = blastx_family WHERE state isnull AND blastn_family LIKE ""; SET final_genus = blastx_genus WHERE state isnull AND blastn_genus LIKE ""; SET final_species = blastx_species WHERE state isnull AND blastn_species LIKE ""; SET final_domain = blastn_domain WHERE state isnull AND blastx_domain LIKE ""; SET final_phylum = blastn_phylum WHERE state isnull AND blastx_phylum LIKE ""; SET final_class = blastn_class

18 WHERE state isnull AND blastx_class LIKE ""; SET final_order = blastn_order WHERE state isnull AND blastx_order LIKE ""; SET final_family = blastn_family WHERE state isnull AND blastx_family LIKE ""; SET final_genus = blastn_genus WHERE state isnull AND blastx_genus LIKE ""; SET final_species = blastn_species WHERE state isnull AND blastx_species LIKE ""; SET state = "assigned" WHERE state isnull AND final_domain not null; d) Contigs that showed hits in both homology searches but were assigned to a different taxon in each, were considered as not assigned with the following command: SET state = "not assigned" WHERE state isnull
, and Consejo Nacional")
19 e) We are finally ready to browse and analyse the updated taxonomy table. For this, go to the Browse Data leaf: Acknowledgements: This research was supported by Agencia Nacional de Promoción Científica y Tecnológica (PICT PRH 112 and PICT CABBIO 3632), and Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) (PIP 0294) grants to CBM. CBM is a member of the CONICET research career. GR and JMC are the recipients of CONICET fellowships. Supplementary material: Background: This workflow was developed to analyse total RNA extracted from insects. Total RNA was submitted to a reverse transcription and polymerase chain reaction sequence-independent amplification procedure, which generated an unbiased random collection of transcripts in which the vast majority corresponded to the host [11 13]. The objective of these studies was to differentiate and determine the host transcriptomic and associated metatranscriptomic profiles of the sample. For this reason, we included all the
20 latest insect genome sequences in our local database to identify host sequences, as well as the non-redundant (nucleotide and protein) and 16S microbial databases, to identify the metatranscriptome. Due to the fact that we observed many differences in the BLASTN and BLASTX results, we developed this workflow to be able to resolve these inconsistencies. It must be noted that, even though this workflow was originally developed to analyse an unbiased collection of high-throughput metatranscriptomic sequences, it has since been used to analyse high-throughput metagenomic shotgun sequences, and works seamlessly. Previous versions of the workflow: Here we mention two previous versions of the workflow, which are described in chronological order to highlight the adjustments and upgrades that led to the final version presented here. In every case, the objective was to be able to observe, compare, and so determine, the correct result for each sequence (and thus for each taxon). Ideally, the results from each homology search would be readily available and visible for each sequence in a table, and the two previous versions of this workflow show how the procedure evolved towards this. I) Workflow designed to analyse total RNA extracted from whole insects (Lutzomyia longipalpis) [12,13]. II) Workflow designed to analyse total RNA extracted from insect guts (Spodoptera frugiperda) [11]. I) Workflow designed to analyse total RNA extracted from whole insects (Lutzomyia longipalpis) [12,13]: Main features: Results were classified and sorted in excel spreadsheets using various custom scripts (written in Mathematica), and then manually combined and compared. The taxonomic and functional profile of the sample was defined on the basis of the different homology search results (and BLAST2GO results, partially) after analysing and curing the spreadsheets manually. MEGAN was not used; No formal criss-crossing was performed. Pros: It was possible to manually combine homology results in a spreadsheet (and so the results from each homology search for each sequence were combined and visible in a single table). Cons: A lot of manual curation required; Very time-consuming; Custom scripts had to be modified each time the format of the homology search outputs differed (however minimal the differences) Workflow: A preliminary version of the L. longipalpis genome (Llon v1.0 contigs, here referred to as Llon_contigs) was downloaded from the Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC) website ( Additionally, five databases were downloaded locally: - non-redundant nucleotide sequences (nt) (NCBI), - 16S ribosomal RNA sequences (Bacteria and Archaea) (16S) (NCBI), - non-human, non-mouse ESTs (est-others) (NCBI), - non-redundant protein sequences (nr) (NCBI), and - UniProt Knowledgebase (uniprotkb), consisting of the Swiss-Prot Protein Knowledgebase (fully annotated curated entries) and the TrEMBL Protein Sequence Database (computer-generated entries enriched with automated classification and annotation) (uniprotkb = uniprot_sprot + uniprot_trembl). In a first stage, sequences from Llon_contigs, nt and 16S were combined in a single database (DB:Llon_contigs+nt+16S) using the appropriate BLAST+ applications (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/latest/). Trimmed reads were compared to this combined database using BLASTN (nucleotide homology) [3], with a 1e-50 cutoff E-value (Figure S1). Reads which mapped to Llon_contigs by sequence homology (here referred to as mapped-reads) were then separated from those that showed homology to nt16s (here referred to as read-nt16s), and an accurate record was kept of which Llon_contig each mapped-read showed homology to, using custom applications written in Mathematica (Wolfram Mathematica 7; available upon request) (Figure S1). Results from the first stage of analysis were organised in three datasets: mapped-reads (reads that mapped to Llon_contigs by sequence homology), read-nt16s (reads that showed homology to nt16s) and no hits (reads that returned no significant hits). Results from the read-nt16s dataset were then classified using custom applications written in Mathematica (Wolfram Mathematica 7; available upon request) and those read-nt16s that did not show homology to insect rdna or taxa other than insects, were selected and separated from the rest to be further analysed (Figure S1). In a second stage, database sequences from nt and 16S were combined in a single database (DB:nt16S) using the appropriate BLAST+ applications (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/latest/), and the mapped-reads dataset was compared to DB:nt16S using BLASTN [3] with a 1e-50 cutoff E-value. After analysing the results of this homology search, it was observed that mapped-reads which in the first stage showed homology to Llon_contigs 15379, 23694, 25834, 27903, and 9281, after BLASTN against DB:nt16S, overall showed homology to insect rdna (these were the only Llon_contigs that were partially annotated after blasting their corresponding mapped-reads against DB:nt16S, i.e. the second stage of analysis), and mapped-
.")
21 reads that in the first stage showed homology to Llon_contig 31202, after BLASTN against DB:nt16S, overall showed no significant hits. Therefore, these mapped-reads were excluded from the third stage of analysis (Figure S1). In the third stage of analysis, all mapped-reads excluding the aforementioned ones (a total of 4910 mapped-reads considering all four samples) and the selected read-nt16s (i.e. those that did not show homology to insect rdna or taxa other than insects, see above), were then compared separately to three databases: DB:est-others using BLASTN [3] with a 1e-50 cutoff E-value, and DB:nr and DB:uniprotKB using BLASTX [7] with a 1e-6 cutoff E-value; these mapped-reads and read-nt16s were also analysed and annotated using Blast2GO [14] (Figure S1). Hits from all four databases and Blast2GO results were compared and individually revised and confirmed, and only those which showed unequivocal results were included in the final analysis. Putative functional assignment and categorisation for each selected mapped-read and read-nt16s was individually and manually revised and assigned on the basis of BLASTX results (mainly DB:uniprotKB) and Blast2GO annotation. Figure S1. Sequence analysis workflow. This figure shows an overview of the rationale supporting the analysis of the reads and summarises the different steps that were followed. 1) Indicates the first stage of analysis, in which all the reads were blasted against DB:Llon_contigs+nt+16S. Results were classified in three datasets: reads that mapped to Llon_contigs by sequence homology (mapped-reads); reads that showed homology to nt16s (read-nt16s); and reads that returned no significant hits (no hits) (see text for details). 2) Indicates the second stage of analysis, in which the mapped-reads dataset was blasted against DB:nt16S (see text for details). 3) Indicates the third stage of analysis, in which selected reads (mapped-reads and read-nt16s) were separately blasted against three databases (est-others, nr and uniprotkb) and annotated using Blast2GO (see text for details). The selected mapped-reads (4910) excluded mapped-reads 15379, 23694, 25834, 27903, and 9281, which showed homology to insect rdna after BLASTN against DB:nt16S, and contig 31202, with unknown function after BLASTN against DB:nt16S (see text for details). The selected read-nt16s (70) included read-nt16s that did not show homology to either insect rdna or taxa other than insects (see text for details). II) Workflow designed to analyse total RNA extracted from insect guts (S. frugiperda) [11]: Main features: MEGAN was used to classify and sort the homology results, in place of the various custom scripts that were used for this same purpose in the previous workflow; Consequently, results were not classified and sorted in excel spreadsheets; The taxonomic and functional profile of the sample was determined based on the assumption that the sequences that correspond to a certain taxon are composed of (Figure S2): a) sequences that were assigned to the same taxon by both homology searches (1 in Figure S2), plus b) sequences that were assigned to that taxon by one of the homology searches but returned no hits in the other one, and vice versa (2 and 3 in Figure S2);
22 For this, the information from both homology searches for each taxon was extracted using one of MEGAN s features, and a custom script (written in Mathematica) was used to intersect this information to identify all the reads that unequivocally corresponded to a certain taxon, and then combine them in a single file. Formal criss-crossing was performed using a custom script (written in Mathematica). Pros: MEGAN greatly simplifies the classification, sorting and manipulation of homology search results (compared to the custom scripts that were used in the previous workflow); Custom scripts were only used to intersect the data. Cons: Homology results were not combined in a spreadsheet and it was thus difficult to visualise the information for each sequence. A lot of manual curation was still required; Time-consuming. Workflow: The following NCBI databases were downloaded locally (ftp://ftp.ncbi.nlm.nih.gov/blast/db/): - Non-redundant nucleotide sequence (nt) (NCBI), - 16S ribosomal RNA sequences (Bacteria and Archaea) (16S) (NCBI), - sequences from lepidopteran whole genome shotgun projects completed at the time of the analysis (Lep) (NCBI), and - non-redundant protein sequence (nr) (NCBI). Sequences from nt, 16S, and Lep, were then combined in a single database (DB:nt+16S+Lep) using the appropriate BLAST+ applications (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/latest/). Trimmed reads were compared to this combined database (DB:nt+16S+Lep) using BLASTN (BN) (nucleotide homology) [3], with a 1e-50 cutoff E-value. Rapsearch2 (RS) [15], which achieves more than 90X acceleration when compared to BLASTX, was used for the protein similarity search against DB:nr with a 1e-7 cutoff E-value. MEGAN (MEtaGenome ANalyzer) [2] was downloaded locally ( and used to analyse both homology search results. A custom script (written in Mathematica) was used to compare the results from the different analyses (see below). This script not only enabled the comparison of the results, but also generated fasta files of the reads that were found in common, and of the reads that were found in one group but not the other, and vice versa. Figure S2 shows an overview of the rationale supporting the sequence analysis workflow used in this study, summarising the different steps that were followed to intersect the information from both analyses and so be able to define the unequivocal taxonomic and functional profile of the sample, based on the assumption that the sequences that correspond to a certain taxon are composed of: a) those sequences that were assigned to the same taxon by both homology searches (1 in Figure S2), plus b) those sequences that were assigned to that taxon by one of the homology searches but returned no hits in the other one, and vice versa (2 and 3 in Figure S2). General workflow: Trimmed reads were submitted to homology searches against DB:nt+16S+Lep and DB:nr using BLASTN (BN) and RAPSearch2 (RS), respectively. The homology search results were analysed with MEGAN and a detailed comparison was then performed in the following way (Figure S2): 1) For each major group, reads were extracted using the appropriate MEGAN tool, and were named reads-nr or reads-nt depending on the homology search they came from (RS or BN, respectively). 2) Reads-nr and reads-nt from the same group (for e.g., Bacteria; Figure S3) were then compared using a custom script (written in Mathematica), and those reads that were found in common were directly assigned to that group (1 in Figures S2 and S3). 3) To retrieve the reads that were assigned to a certain taxon by one of the homology searches but returned no hits in the other one, and vice versa (2 and 3 in Figures S2 and S3), two different analyses were performed (which produced the same end result): First analysis: 3) a. i. Using the custom script, reads-nr were compared with all the reads that retrieved no hits in the global BN analysis (Total no hits BN in Figures S2 and S3), and reads-nt were compared with all the reads that retrieved no hits in the global RS analysis (Total no hits RS in Figures S2 and S3). 3) a. ii. Reads that were found in common (i.e., reads-nr that had no hits in BN, 2 in Figures S2 and S3, and reads-nt that had no hits in RS, 3 in Figures S2 and S3), were also assigned to the taxonomical group that was being analysed (for e.g., Bacteria; Figure S3). 3) a. iii. Reads that showed hits to different taxa in the RS and BN analyses (for e.g., Bacteria in one analysis and Fungi in the other), were considered not assigned. The only exception to this was Arthropoda, because various taxonomical groups that appeared in the RS analysis did not figure in the BN analysis, and when the reads for these groups were extracted and separately analysed with BN, they showed unequivocal homology to Arthropoda. For this reason, they were subsequently assigned to Arthropoda. Second analysis:
23 3) b. i. All the reads that retrieved no hits in the global RS (nohits-nr) and BN (nohits-nt) analyses, were extracted using the appropriate MEGAN tool, and compared using the custom script. Here again, reads that were found in common were unquestionably assigned as no hits. 3) b. ii. On the other hand, reads that only retrieved no hits in either the RS or BN analyses, were submitted to BN and RS homology searches, respectively, and the results were subsequently analysed with MEGAN. 3) b. iii. If the results in one of these searches included reads that were assigned to a certain taxon (for e.g., Bacteria), they were assigned to that taxon (2 and 3 in Figure S3). 4) After this criss-crossing, reads belonging to the taxonomical group being analysed were combined in a single multifasta ( total reads = in Figures S2 and S3), submitted to RS and BN analysis, and these results were then processed with MEGAN. In this way, since all the reads that unequivocally corresponded to a certain taxon were combined in a single file, it was possible to discriminate and characterise the taxonomic and functional profile of the sample. Figure S2. General workflow to define the unequivocal taxonomical and functional profile of a sample. RS = RapSearch2; BN = BLASTN;.aln = RS file extension;.txt = BN file extension;.rma = MEGAN file extension; 1 = sequences that were assigned to taxon X by both homology searches; 2 = sequences that were assigned to taxon X by RS but returned no hits in BN; 3 = sequences that were assigned to taxon X by BN but returned no hits in RS.
24 Figure S3. Example of how the workflow was used to define the unequivocal taxonomical and functional profile of Bacteria in the sample. RS = RapSearch2; BN = BLASTN;.aln = RS file extension; reads-nt = reads that showed homology to Bacteria in the BN analysis; reads-nr = reads that showed homology to Bacteria in the RS analysis; nohits-nt = reads that retrieved no results in the BN analysis; nohits-nr = reads that retrieved no results in the RS analysis;.txt = BN file extension;.rma = MEGAN file extension; 1 = sequences that were assigned to taxon X by both homology searches; 2 = sequences that were assigned to taxon X by RS but returned no hits in BN; 3 = sequences that were assigned to taxon X by BN but returned no hits in RS.
25 *References: [1] J.C. Wooley, A. Godzik, I. Friedberg, A primer on metagenomics, PLoS Comput. Biol. (2010). doi: /journal.pcbi [2] D.H. Huson, A.F. Auch, J. Qi, S.C. Schuster, MEGAN analysis of metagenomic data, Genome Res. (2007). doi: /gr [3] S.F. Altschul, W. Gish, W. Miller, E.W. Myers, D.J. Lipman, Basic local alignment search tool, J. Mol. Biol. 215 (1990) [4] M. Kim, K.-H. Lee, S.-W. Yoon, B.-S. Kim, J. Chun, H. Yi, Analytical Tools and Databases for Metagenomics in the Next- Generation Sequencing Era, Genomics Inform. (2013). doi: /gi [5] V. Aguiar-Pulido, W. Huang, V. Suarez-Ulloa, T. Cickovski, K. Mathee, G. Narasimhan, Metagenomics, metatranscriptomics, and metabolomics approaches for microbiome analysis, Evol. Bioinforma. (2016). doi: /ebo.s [6] D.H. Huson, S. Mitra, H.J. Ruscheweyh, N. Weber, S.C. Schuster, Integrative analysis of environmental sequences using MEGAN4, Genome Res. 21 (2011) doi:gr [pii] /gr [7] S.F. Altschul, T.L. Madden, A.A. Schäffer, J. Zhang, Z. Zhang, W. Miller, D.J. Lipman, Gapped BLAST and PSI-BLAST: A new generation of protein database search programs, Nucleic Acids Res. 25 (1997) doi: /nar/ [8] R. Overbeek, T. Begley, R.M. Butler, J. V Choudhuri, H.Y. Chuang, M. Cohoon, V. de Crecy-Lagard, N. Diaz, T. Disz, R. Edwards, M. Fonstein, E.D. Frank, S. Gerdes, E.M. Glass, A. Goesmann, A. Hanson, D. Iwata-Reuyl, R. Jensen, N. Jamshidi, L. Krause, M. Kubal, N. Larsen, B. Linke, A.C. McHardy, F. Meyer, H. Neuweger, G. Olsen, R. Olson, A. Osterman, V. Portnoy, G.D. Pusch, D.A. Rodionov, C. Ruckert, J. Steiner, R. Stevens, I. Thiele, O. Vassieva, Y. Ye, O. Zagnitko, V. Vonstein, The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes, Nucleic Acids Res. 33 (2005) doi:33/17/5691 [pii] /nar/gki866. [9] R.L. Tatusov, The COG database: a tool for genome-scale analysis of protein functions and evolution, Nucleic Acids Res. (2000). doi: /nar/ [10] H. Ogata, S. Goto, K. Sato, W. Fujibuchi, H. Bono, M. Kanehisa, KEGG: Kyoto encyclopedia of genes and genomes, Nucleic Acids Res. (1999). doi: /nar/ [11] C.B. McCarthy, N.A. Cabrera, E.G. Virla, Metatranscriptomic analysis of larval guts from field-collected and laboratoryreared Spodoptera frugiperda from the South American subtropical region, Genome Announc. 3 (2015) e [12] C.B. McCarthy, M.S. Santini, P.F.P. Pimenta, L. a. Diambra, First Comparative Transcriptomic Analysis of Wild Adult Male and Female Lutzomyia longipalpis, Vector of Visceral Leishmaniasis, PLoS One. 8 (2013) e doi: /journal.pone [13] C.B. McCarthy, L. a Diambra, R. V Rivera Pomar, Metagenomic analysis of taxa associated with Lutzomyia longipalpis, vector of visceral leishmaniasis, using an unbiased high-throughput approach., PLoS Negl. Trop. Dis. 5 (2011) e1304. doi: /journal.pntd [14] A. Conesa, S. Gotz, J.M. Garcia-Gomez, J. Terol, M. Talon, M. Robles, Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research, Bioinformatics. 21 (2005) doi:bti610 [pii] /bioinformatics/bti610. [15] Y. Zhao, H. Tang, Y. Ye, RAPSearch2: A fast and memory-efficient protein similarity search tool for next-generation sequencing data, Bioinformatics. 28 (2012) doi: /bioinformatics/btr595.
BLAST. Varieties of BLAST
 BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
BLAST Database Searching. BME 110: CompBio Tools Todd Lowe April 8, 2010
 BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
SUPPLEMENTARY INFORMATION
 Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Microbial analysis with STAMP
 Microbial analysis with STAMP Conor Meehan cmeehan@itg.be A quick aside on who I am Tangents already! Who I am A postdoc at the Institute of Tropical Medicine in Antwerp, Belgium Mycobacteria evolution
Microbial analysis with STAMP Conor Meehan cmeehan@itg.be A quick aside on who I am Tangents already! Who I am A postdoc at the Institute of Tropical Medicine in Antwerp, Belgium Mycobacteria evolution
Introduction to Bioinformatics Online Course: IBT
 Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
ATLAS of Biochemistry
 ATLAS of Biochemistry USER GUIDE http://lcsb-databases.epfl.ch/atlas/ CONTENT 1 2 3 GET STARTED Create your user account NAVIGATE Curated KEGG reactions ATLAS reactions Pathways Maps USE IT! Fill a gap
ATLAS of Biochemistry USER GUIDE http://lcsb-databases.epfl.ch/atlas/ CONTENT 1 2 3 GET STARTED Create your user account NAVIGATE Curated KEGG reactions ATLAS reactions Pathways Maps USE IT! Fill a gap
SUPPLEMENTARY INFORMATION
 Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Biology 559R: Introduction to Phylogenetic Comparative Methods Topics for this week:
 Biology 559R: Introduction to Phylogenetic Comparative Methods Topics for this week: Course general information About the course Course objectives Comparative methods: An overview R as language: uses and
Biology 559R: Introduction to Phylogenetic Comparative Methods Topics for this week: Course general information About the course Course objectives Comparative methods: An overview R as language: uses and
Hands-On Nine The PAX6 Gene and Protein
 Hands-On Nine The PAX6 Gene and Protein Main Purpose of Hands-On Activity: Using bioinformatics tools to examine the sequences, homology, and disease relevance of the Pax6: a master gene of eye formation.
Hands-On Nine The PAX6 Gene and Protein Main Purpose of Hands-On Activity: Using bioinformatics tools to examine the sequences, homology, and disease relevance of the Pax6: a master gene of eye formation.
Bioinformatics. Dept. of Computational Biology & Bioinformatics
 Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Comparing whole genomes
 BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
MiGA: The Microbial Genome Atlas
 December 12 th 2017 MiGA: The Microbial Genome Atlas Jim Cole Center for Microbial Ecology Dept. of Plant, Soil & Microbial Sciences Michigan State University East Lansing, Michigan U.S.A. Where I m From
December 12 th 2017 MiGA: The Microbial Genome Atlas Jim Cole Center for Microbial Ecology Dept. of Plant, Soil & Microbial Sciences Michigan State University East Lansing, Michigan U.S.A. Where I m From
Mathangi Thiagarajan Rice Genome Annotation Workshop May 23rd, 2007
 -2 Transcript Alignment Assembly and Automated Gene Structure Improvements Using PASA-2 Mathangi Thiagarajan mathangi@jcvi.org Rice Genome Annotation Workshop May 23rd, 2007 About PASA PASA is an open
-2 Transcript Alignment Assembly and Automated Gene Structure Improvements Using PASA-2 Mathangi Thiagarajan mathangi@jcvi.org Rice Genome Annotation Workshop May 23rd, 2007 About PASA PASA is an open
Microbiome: 16S rrna Sequencing 3/30/2018
 Microbiome: 16S rrna Sequencing 3/30/2018 Skills from Previous Lectures Central Dogma of Biology Lecture 3: Genetics and Genomics Lecture 4: Microarrays Lecture 12: ChIP-Seq Phylogenetics Lecture 13: Phylogenetics
Microbiome: 16S rrna Sequencing 3/30/2018 Skills from Previous Lectures Central Dogma of Biology Lecture 3: Genetics and Genomics Lecture 4: Microarrays Lecture 12: ChIP-Seq Phylogenetics Lecture 13: Phylogenetics
Supplementary Materials for R3P-Loc Web-server
 Supplementary Materials for R3P-Loc Web-server Shibiao Wan and Man-Wai Mak email: shibiao.wan@connect.polyu.hk, enmwmak@polyu.edu.hk June 2014 Back to R3P-Loc Server Contents 1 Introduction to R3P-Loc
Supplementary Materials for R3P-Loc Web-server Shibiao Wan and Man-Wai Mak email: shibiao.wan@connect.polyu.hk, enmwmak@polyu.edu.hk June 2014 Back to R3P-Loc Server Contents 1 Introduction to R3P-Loc
Centrifuge: rapid and sensitive classification of metagenomic sequences
 Centrifuge: rapid and sensitive classification of metagenomic sequences Daehwan Kim, Li Song, Florian P. Breitwieser, and Steven L. Salzberg Supplementary Material Supplementary Table 1 Supplementary Note
Centrifuge: rapid and sensitive classification of metagenomic sequences Daehwan Kim, Li Song, Florian P. Breitwieser, and Steven L. Salzberg Supplementary Material Supplementary Table 1 Supplementary Note
Genome Annotation. Bioinformatics and Computational Biology. Genome sequencing Assembly. Gene prediction. Protein targeting.
 Genome Annotation Bioinformatics and Computational Biology Genome Annotation Frank Oliver Glöckner 1 Genome Analysis Roadmap Genome sequencing Assembly Gene prediction Protein targeting trna prediction
Genome Annotation Bioinformatics and Computational Biology Genome Annotation Frank Oliver Glöckner 1 Genome Analysis Roadmap Genome sequencing Assembly Gene prediction Protein targeting trna prediction
Taxonomical Classification using:
 Taxonomical Classification using: Extracting ecological signal from noise: introduction to tools for the analysis of NGS data from microbial communities Bergen, April 19-20 2012 INTRODUCTION Taxonomical
Taxonomical Classification using: Extracting ecological signal from noise: introduction to tools for the analysis of NGS data from microbial communities Bergen, April 19-20 2012 INTRODUCTION Taxonomical
OECD QSAR Toolbox v.4.1. Step-by-step example for building QSAR model
 OECD QSAR Toolbox v.4.1 Step-by-step example for building QSAR model Background Objectives The exercise Workflow of the exercise Outlook 2 Background This is a step-by-step presentation designed to take
OECD QSAR Toolbox v.4.1 Step-by-step example for building QSAR model Background Objectives The exercise Workflow of the exercise Outlook 2 Background This is a step-by-step presentation designed to take
08/21/2017 BLAST. Multiple Sequence Alignments: Clustal Omega
 BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
Title ghost-tree: creating hybrid-gene phylogenetic trees for diversity analyses
 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 Title ghost-tree: creating hybrid-gene phylogenetic trees for diversity analyses
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 Title ghost-tree: creating hybrid-gene phylogenetic trees for diversity analyses
PGA: A Program for Genome Annotation by Comparative Analysis of. Maximum Likelihood Phylogenies of Genes and Species
 PGA: A Program for Genome Annotation by Comparative Analysis of Maximum Likelihood Phylogenies of Genes and Species Paulo Bandiera-Paiva 1 and Marcelo R.S. Briones 2 1 Departmento de Informática em Saúde
PGA: A Program for Genome Annotation by Comparative Analysis of Maximum Likelihood Phylogenies of Genes and Species Paulo Bandiera-Paiva 1 and Marcelo R.S. Briones 2 1 Departmento de Informática em Saúde
Introduction to Bioinformatics
 Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Grundlagen der Bioinformatik Summer semester Lecturer: Prof. Daniel Huson
 Grundlagen der Bioinformatik, SS 10, D. Huson, April 12, 2010 1 1 Introduction Grundlagen der Bioinformatik Summer semester 2010 Lecturer: Prof. Daniel Huson Office hours: Thursdays 17-18h (Sand 14, C310a)
Grundlagen der Bioinformatik, SS 10, D. Huson, April 12, 2010 1 1 Introduction Grundlagen der Bioinformatik Summer semester 2010 Lecturer: Prof. Daniel Huson Office hours: Thursdays 17-18h (Sand 14, C310a)
OECD QSAR Toolbox v.3.2. Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding
 OECD QSAR Toolbox v.3.2 Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding Outlook Background Objectives Specific Aims The exercise Workflow
OECD QSAR Toolbox v.3.2 Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding Outlook Background Objectives Specific Aims The exercise Workflow
GEP Annotation Report
 GEP Annotation Report Note: For each gene described in this annotation report, you should also prepare the corresponding GFF, transcript and peptide sequence files as part of your submission. Student name:
GEP Annotation Report Note: For each gene described in this annotation report, you should also prepare the corresponding GFF, transcript and peptide sequence files as part of your submission. Student name:
Lecture 2. The Blast2GO annotation framework
 Lecture 2 The Blast2GO annotation framework Annotation steps Modulation of annotation intensity Export/Import Functions Sequence Selection Additional Tools Functional assignment Annotation Transference
Lecture 2 The Blast2GO annotation framework Annotation steps Modulation of annotation intensity Export/Import Functions Sequence Selection Additional Tools Functional assignment Annotation Transference
Heuristic Alignment and Searching
 3/28/2012 Types of alignments Global Alignment Each letter of each sequence is aligned to a letter or a gap (e.g., Needleman-Wunsch). Local Alignment An optimal pair of subsequences is taken from the two
3/28/2012 Types of alignments Global Alignment Each letter of each sequence is aligned to a letter or a gap (e.g., Needleman-Wunsch). Local Alignment An optimal pair of subsequences is taken from the two
Small RNA in rice genome
 Vol. 45 No. 5 SCIENCE IN CHINA (Series C) October 2002 Small RNA in rice genome WANG Kai ( 1, ZHU Xiaopeng ( 2, ZHONG Lan ( 1,3 & CHEN Runsheng ( 1,2 1. Beijing Genomics Institute/Center of Genomics and
Vol. 45 No. 5 SCIENCE IN CHINA (Series C) October 2002 Small RNA in rice genome WANG Kai ( 1, ZHU Xiaopeng ( 2, ZHONG Lan ( 1,3 & CHEN Runsheng ( 1,2 1. Beijing Genomics Institute/Center of Genomics and
Investigation 3: Comparing DNA Sequences to Understand Evolutionary Relationships with BLAST
 Investigation 3: Comparing DNA Sequences to Understand Evolutionary Relationships with BLAST Introduction Bioinformatics is a powerful tool which can be used to determine evolutionary relationships and
Investigation 3: Comparing DNA Sequences to Understand Evolutionary Relationships with BLAST Introduction Bioinformatics is a powerful tool which can be used to determine evolutionary relationships and
OECD QSAR Toolbox v.3.3. Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding
 OECD QSAR Toolbox v.3.3 Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding Outlook Background Objectives Specific Aims The exercise Workflow
OECD QSAR Toolbox v.3.3 Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding Outlook Background Objectives Specific Aims The exercise Workflow
Supplementary Materials for mplr-loc Web-server
 Supplementary Materials for mplr-loc Web-server Shibiao Wan and Man-Wai Mak email: shibiao.wan@connect.polyu.hk, enmwmak@polyu.edu.hk June 2014 Back to mplr-loc Server Contents 1 Introduction to mplr-loc
Supplementary Materials for mplr-loc Web-server Shibiao Wan and Man-Wai Mak email: shibiao.wan@connect.polyu.hk, enmwmak@polyu.edu.hk June 2014 Back to mplr-loc Server Contents 1 Introduction to mplr-loc
Assigning Taxonomy to Marker Genes. Susan Huse Brown University August 7, 2014
 Assigning Taxonomy to Marker Genes Susan Huse Brown University August 7, 2014 In a nutshell Taxonomy is assigned by comparing your DNA sequences against a database of DNA sequences from known taxa Marker
Assigning Taxonomy to Marker Genes Susan Huse Brown University August 7, 2014 In a nutshell Taxonomy is assigned by comparing your DNA sequences against a database of DNA sequences from known taxa Marker
CS612 - Algorithms in Bioinformatics
 Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
Search for the Gulf of Carpentaria in the remap search bar:
 This tutorial is aimed at getting you started with making maps in Remap (). In this tutorial we are going to develop a simple classification of mangroves in northern Australia. Before getting started with
This tutorial is aimed at getting you started with making maps in Remap (). In this tutorial we are going to develop a simple classification of mangroves in northern Australia. Before getting started with
Tiffany Samaroo MB&B 452a December 8, Take Home Final. Topic 1
 Tiffany Samaroo MB&B 452a December 8, 2003 Take Home Final Topic 1 Prior to 1970, protein and DNA sequence alignment was limited to visual comparison. This was a very tedious process; even proteins with
Tiffany Samaroo MB&B 452a December 8, 2003 Take Home Final Topic 1 Prior to 1970, protein and DNA sequence alignment was limited to visual comparison. This was a very tedious process; even proteins with
Supplemental Materials
 JOURNAL OF MICROBIOLOGY & BIOLOGY EDUCATION, May 2013, p. 107-109 DOI: http://dx.doi.org/10.1128/jmbe.v14i1.496 Supplemental Materials for Engaging Students in a Bioinformatics Activity to Introduce Gene
JOURNAL OF MICROBIOLOGY & BIOLOGY EDUCATION, May 2013, p. 107-109 DOI: http://dx.doi.org/10.1128/jmbe.v14i1.496 Supplemental Materials for Engaging Students in a Bioinformatics Activity to Introduce Gene
ST-Links. SpatialKit. Version 3.0.x. For ArcMap. ArcMap Extension for Directly Connecting to Spatial Databases. ST-Links Corporation.
 ST-Links SpatialKit For ArcMap Version 3.0.x ArcMap Extension for Directly Connecting to Spatial Databases ST-Links Corporation www.st-links.com 2012 Contents Introduction... 3 Installation... 3 Database
ST-Links SpatialKit For ArcMap Version 3.0.x ArcMap Extension for Directly Connecting to Spatial Databases ST-Links Corporation www.st-links.com 2012 Contents Introduction... 3 Installation... 3 Database
OECD QSAR Toolbox v.3.4. Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding
 OECD QSAR Toolbox v.3.4 Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding Outlook Background Objectives Specific Aims The exercise Workflow
OECD QSAR Toolbox v.3.4 Step-by-step example of how to build and evaluate a category based on mechanism of action with protein and DNA binding Outlook Background Objectives Specific Aims The exercise Workflow
Journal of Proteomics & Bioinformatics - Open Access
 Abstract Methodology for Phylogenetic Tree Construction Kudipudi Srinivas 2, Allam Appa Rao 1, GR Sridhar 3, Srinubabu Gedela 1* 1 International Center for Bioinformatics & Center for Biotechnology, Andhra
Abstract Methodology for Phylogenetic Tree Construction Kudipudi Srinivas 2, Allam Appa Rao 1, GR Sridhar 3, Srinubabu Gedela 1* 1 International Center for Bioinformatics & Center for Biotechnology, Andhra
Reaxys Pipeline Pilot Components Installation and User Guide
 1 1 Reaxys Pipeline Pilot components for Pipeline Pilot 9.5 Reaxys Pipeline Pilot Components Installation and User Guide Version 1.0 2 Introduction The Reaxys and Reaxys Medicinal Chemistry Application
1 1 Reaxys Pipeline Pilot components for Pipeline Pilot 9.5 Reaxys Pipeline Pilot Components Installation and User Guide Version 1.0 2 Introduction The Reaxys and Reaxys Medicinal Chemistry Application
Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis
 Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis Technical Overview Introduction Metabolomics studies measure the relative abundance of metabolites
Agilent MassHunter Profinder: Solving the Challenge of Isotopologue Extraction for Qualitative Flux Analysis Technical Overview Introduction Metabolomics studies measure the relative abundance of metabolites
Genome Annotation. Qi Sun Bioinformatics Facility Cornell University
 Genome Annotation Qi Sun Bioinformatics Facility Cornell University Some basic bioinformatics tools BLAST PSI-BLAST - Position-Specific Scoring Matrix HMM - Hidden Markov Model NCBI BLAST How does BLAST
Genome Annotation Qi Sun Bioinformatics Facility Cornell University Some basic bioinformatics tools BLAST PSI-BLAST - Position-Specific Scoring Matrix HMM - Hidden Markov Model NCBI BLAST How does BLAST
Last updated: Copyright
 Last updated: 2012-08-20 Copyright 2004-2012 plabel (v2.4) User s Manual by Bioinformatics Group, Institute of Computing Technology, Chinese Academy of Sciences Tel: 86-10-62601016 Email: zhangkun01@ict.ac.cn,
Last updated: 2012-08-20 Copyright 2004-2012 plabel (v2.4) User s Manual by Bioinformatics Group, Institute of Computing Technology, Chinese Academy of Sciences Tel: 86-10-62601016 Email: zhangkun01@ict.ac.cn,
DATA ACQUISITION FROM BIO-DATABASES AND BLAST. Natapol Pornputtapong 18 January 2018
 DATA ACQUISITION FROM BIO-DATABASES AND BLAST Natapol Pornputtapong 18 January 2018 DATABASE Collections of data To share multi-user interface To prevent data loss To make sure to get the right things
DATA ACQUISITION FROM BIO-DATABASES AND BLAST Natapol Pornputtapong 18 January 2018 DATABASE Collections of data To share multi-user interface To prevent data loss To make sure to get the right things
Homology. and. Information Gathering and Domain Annotation for Proteins
 Homology and Information Gathering and Domain Annotation for Proteins Outline WHAT IS HOMOLOGY? HOW TO GATHER KNOWN PROTEIN INFORMATION? HOW TO ANNOTATE PROTEIN DOMAINS? EXAMPLES AND EXERCISES Homology
Homology and Information Gathering and Domain Annotation for Proteins Outline WHAT IS HOMOLOGY? HOW TO GATHER KNOWN PROTEIN INFORMATION? HOW TO ANNOTATE PROTEIN DOMAINS? EXAMPLES AND EXERCISES Homology
Bacterial Communities in Women with Bacterial Vaginosis: High Resolution Phylogenetic Analyses Reveal Relationships of Microbiota to Clinical Criteria
 Bacterial Communities in Women with Bacterial Vaginosis: High Resolution Phylogenetic Analyses Reveal Relationships of Microbiota to Clinical Criteria Seminar presentation Pierre Barbera Supervised by:
Bacterial Communities in Women with Bacterial Vaginosis: High Resolution Phylogenetic Analyses Reveal Relationships of Microbiota to Clinical Criteria Seminar presentation Pierre Barbera Supervised by:
Photometry of Supernovae with Makali i
 Photometry of Supernovae with Makali i How to perform photometry specifically on supernovae targets using the free image processing software, Makali i This worksheet describes how to use photometry to
Photometry of Supernovae with Makali i How to perform photometry specifically on supernovae targets using the free image processing software, Makali i This worksheet describes how to use photometry to
Grundlagen der Bioinformatik, SS 08, D. Huson, May 2,
 Grundlagen der Bioinformatik, SS 08, D. Huson, May 2, 2008 39 5 Blast This lecture is based on the following, which are all recommended reading: R. Merkl, S. Waack: Bioinformatik Interaktiv. Chapter 11.4-11.7
Grundlagen der Bioinformatik, SS 08, D. Huson, May 2, 2008 39 5 Blast This lecture is based on the following, which are all recommended reading: R. Merkl, S. Waack: Bioinformatik Interaktiv. Chapter 11.4-11.7
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
OECD QSAR Toolbox v.3.0
 OECD QSAR Toolbox v.3.0 Step-by-step example of how to categorize an inventory by mechanistic behaviour of the chemicals which it consists Background Objectives Specific Aims Trend analysis The exercise
OECD QSAR Toolbox v.3.0 Step-by-step example of how to categorize an inventory by mechanistic behaviour of the chemicals which it consists Background Objectives Specific Aims Trend analysis The exercise
Compounding insights Thermo Scientific Compound Discoverer Software
 Compounding insights Thermo Scientific Compound Discoverer Software Integrated, complete, toolset solves small-molecule analysis challenges Thermo Scientific Orbitrap mass spectrometers produce information-rich
Compounding insights Thermo Scientific Compound Discoverer Software Integrated, complete, toolset solves small-molecule analysis challenges Thermo Scientific Orbitrap mass spectrometers produce information-rich
A powerful site for all chemists CHOICE CRC Handbook of Chemistry and Physics
 Chemical Databases Online A powerful site for all chemists CHOICE CRC Handbook of Chemistry and Physics Combined Chemical Dictionary Dictionary of Natural Products Dictionary of Organic Dictionary of Drugs
Chemical Databases Online A powerful site for all chemists CHOICE CRC Handbook of Chemistry and Physics Combined Chemical Dictionary Dictionary of Natural Products Dictionary of Organic Dictionary of Drugs
PHYLOGENY AND SYSTEMATICS
 AP BIOLOGY EVOLUTION/HEREDITY UNIT Unit 1 Part 11 Chapter 26 Activity #15 NAME DATE PERIOD PHYLOGENY AND SYSTEMATICS PHYLOGENY Evolutionary history of species or group of related species SYSTEMATICS Study
AP BIOLOGY EVOLUTION/HEREDITY UNIT Unit 1 Part 11 Chapter 26 Activity #15 NAME DATE PERIOD PHYLOGENY AND SYSTEMATICS PHYLOGENY Evolutionary history of species or group of related species SYSTEMATICS Study
OECD QSAR Toolbox v.4.1. Tutorial on how to predict Skin sensitization potential taking into account alert performance
 OECD QSAR Toolbox v.4.1 Tutorial on how to predict Skin sensitization potential taking into account alert performance Outlook Background Objectives Specific Aims Read across and analogue approach The exercise
OECD QSAR Toolbox v.4.1 Tutorial on how to predict Skin sensitization potential taking into account alert performance Outlook Background Objectives Specific Aims Read across and analogue approach The exercise
OECD QSAR Toolbox v.3.3. Predicting acute aquatic toxicity to fish of Dodecanenitrile (CAS ) taking into account tautomerism
 taking into account tautomerism") OECD QSAR Toolbox v.3.3 Predicting acute aquatic toxicity to fish of Dodecanenitrile (CAS 2437-25-4) taking into account tautomerism Outlook Background Objectives The exercise Workflow Save prediction
OECD QSAR Toolbox v.3.3 Predicting acute aquatic toxicity to fish of Dodecanenitrile (CAS 2437-25-4) taking into account tautomerism Outlook Background Objectives The exercise Workflow Save prediction
Bioinformatics tools for phylogeny and visualization. Yanbin Yin
 Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
Bioinformatics Exercises
 Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
Effective Cluster-Based Seed Design for Cross-Species Sequence Comparisons
 Effective Cluster-Based Seed Design for Cross-Species Sequence Comparisons Leming Zhou and Liliana Florea 1 Methods Supplementary Materials 1.1 Cluster-based seed design 1. Determine Homologous Genes.
Effective Cluster-Based Seed Design for Cross-Species Sequence Comparisons Leming Zhou and Liliana Florea 1 Methods Supplementary Materials 1.1 Cluster-based seed design 1. Determine Homologous Genes.
Synteny Portal Documentation
 Synteny Portal Documentation Synteny Portal is a web application portal for visualizing, browsing, searching and building synteny blocks. Synteny Portal provides four main web applications: SynCircos,
Synteny Portal Documentation Synteny Portal is a web application portal for visualizing, browsing, searching and building synteny blocks. Synteny Portal provides four main web applications: SynCircos,
OECD QSAR Toolbox v.4.0. Tutorial on how to predict Skin sensitization potential taking into account alert performance
 OECD QSAR Toolbox v.4.0 Tutorial on how to predict Skin sensitization potential taking into account alert performance Outlook Background Objectives Specific Aims Read across and analogue approach The exercise
OECD QSAR Toolbox v.4.0 Tutorial on how to predict Skin sensitization potential taking into account alert performance Outlook Background Objectives Specific Aims Read across and analogue approach The exercise
Basic Local Alignment Search Tool
 Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
OECD QSAR Toolbox v.3.3. Step-by-step example of how to build a userdefined
 OECD QSAR Toolbox v.3.3 Step-by-step example of how to build a userdefined QSAR Background Objectives The exercise Workflow of the exercise Outlook 2 Background This is a step-by-step presentation designed
OECD QSAR Toolbox v.3.3 Step-by-step example of how to build a userdefined QSAR Background Objectives The exercise Workflow of the exercise Outlook 2 Background This is a step-by-step presentation designed
OECD QSAR Toolbox v.4.1
 OECD QSAR Toolbox v.4.1 Step-by-step example on how to predict the skin sensitisation potential approach of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific
OECD QSAR Toolbox v.4.1 Step-by-step example on how to predict the skin sensitisation potential approach of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific
Single alignment: Substitution Matrix. 16 march 2017
 Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
ProtoNet 4.0: A hierarchical classification of one million protein sequences
 ProtoNet 4.0: A hierarchical classification of one million protein sequences Noam Kaplan 1*, Ori Sasson 2, Uri Inbar 2, Moriah Friedlich 2, Menachem Fromer 2, Hillel Fleischer 2, Elon Portugaly 2, Nathan
ProtoNet 4.0: A hierarchical classification of one million protein sequences Noam Kaplan 1*, Ori Sasson 2, Uri Inbar 2, Moriah Friedlich 2, Menachem Fromer 2, Hillel Fleischer 2, Elon Portugaly 2, Nathan
1 Abstract. 2 Introduction. 3 Requirements. 4 Procedure
 1 Abstract None 2 Introduction The archaeal core set is used in testing the completeness of the archaeal draft genomes. The core set comprises of conserved single copy genes from 25 genomes. Coverage statistic
1 Abstract None 2 Introduction The archaeal core set is used in testing the completeness of the archaeal draft genomes. The core set comprises of conserved single copy genes from 25 genomes. Coverage statistic
COMPARATIVE PATHWAY ANNOTATION WITH PROTEIN-DNA INTERACTION AND OPERON INFORMATION VIA GRAPH TREE DECOMPOSITION
 COMPARATIVE PATHWAY ANNOTATION WITH PROTEIN-DNA INTERACTION AND OPERON INFORMATION VIA GRAPH TREE DECOMPOSITION JIZHEN ZHAO, DONGSHENG CHE AND LIMING CAI Department of Computer Science, University of Georgia,
COMPARATIVE PATHWAY ANNOTATION WITH PROTEIN-DNA INTERACTION AND OPERON INFORMATION VIA GRAPH TREE DECOMPOSITION JIZHEN ZHAO, DONGSHENG CHE AND LIMING CAI Department of Computer Science, University of Georgia,
M E R C E R W I N WA L K T H R O U G H
 H E A L T H W E A L T H C A R E E R WA L K T H R O U G H C L I E N T S O L U T I O N S T E A M T A B L E O F C O N T E N T 1. Login to the Tool 2 2. Published reports... 7 3. Select Results Criteria...
H E A L T H W E A L T H C A R E E R WA L K T H R O U G H C L I E N T S O L U T I O N S T E A M T A B L E O F C O N T E N T 1. Login to the Tool 2 2. Published reports... 7 3. Select Results Criteria...
OECD QSAR Toolbox v.3.3. Step-by-step example of how to categorize an inventory by mechanistic behaviour of the chemicals which it consists
 OECD QSAR Toolbox v.3.3 Step-by-step example of how to categorize an inventory by mechanistic behaviour of the chemicals which it consists Background Objectives Specific Aims Trend analysis The exercise
OECD QSAR Toolbox v.3.3 Step-by-step example of how to categorize an inventory by mechanistic behaviour of the chemicals which it consists Background Objectives Specific Aims Trend analysis The exercise
BIOINFORMATICS: An Introduction
 BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
ncounter PlexSet Data Analysis Guidelines
 ncounter PlexSet Data Analysis Guidelines NanoString Technologies, Inc. 530 airview Ave North Seattle, Washington 98109 USA Telephone: 206.378.6266 888.358.6266 E-mail: info@nanostring.com Molecules That
ncounter PlexSet Data Analysis Guidelines NanoString Technologies, Inc. 530 airview Ave North Seattle, Washington 98109 USA Telephone: 206.378.6266 888.358.6266 E-mail: info@nanostring.com Molecules That
Reaxys Medicinal Chemistry Fact Sheet
 R&D SOLUTIONS FOR PHARMA & LIFE SCIENCES Reaxys Medicinal Chemistry Fact Sheet Essential data for lead identification and optimization Reaxys Medicinal Chemistry empowers early discovery in drug development
R&D SOLUTIONS FOR PHARMA & LIFE SCIENCES Reaxys Medicinal Chemistry Fact Sheet Essential data for lead identification and optimization Reaxys Medicinal Chemistry empowers early discovery in drug development
Browsing Genomic Information with Ensembl Plants
 Browsing Genomic Information with Ensembl Plants Etienne de Villiers, PhD (Adapted from slides by Bert Overduin EMBL-EBI) Outline of workshop Brief introduction to Ensembl Plants History Content Tutorial
Browsing Genomic Information with Ensembl Plants Etienne de Villiers, PhD (Adapted from slides by Bert Overduin EMBL-EBI) Outline of workshop Brief introduction to Ensembl Plants History Content Tutorial
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
OECD QSAR Toolbox v.3.4
 OECD QSAR Toolbox v.3.4 Step-by-step example on how to predict the skin sensitisation potential approach of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific
OECD QSAR Toolbox v.3.4 Step-by-step example on how to predict the skin sensitisation potential approach of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific
GENE ONTOLOGY (GO) Wilver Martínez Martínez Giovanny Silva Rincón
 Wilver Martínez Martínez Giovanny Silva Rincón") GENE ONTOLOGY (GO) Wilver Martínez Martínez Giovanny Silva Rincón What is GO? The Gene Ontology (GO) project is a collaborative effort to address the need for consistent descriptions of gene products in
GENE ONTOLOGY (GO) Wilver Martínez Martínez Giovanny Silva Rincón What is GO? The Gene Ontology (GO) project is a collaborative effort to address the need for consistent descriptions of gene products in
Ensembl focuses on metazoan (animal) genomes. The genomes currently available at the Ensembl site are:
 genomes. The genomes currently available at the Ensembl site are:") Comparative genomics and proteomics Species available Ensembl focuses on metazoan (animal) genomes. The genomes currently available at the Ensembl site are: Vertebrates: human, chimpanzee, mouse, rat,
Comparative genomics and proteomics Species available Ensembl focuses on metazoan (animal) genomes. The genomes currently available at the Ensembl site are: Vertebrates: human, chimpanzee, mouse, rat,
-max_target_seqs: maximum number of targets to report
 Review of exercise 1 tblastn -num_threads 2 -db contig -query DH10B.fasta -out blastout.xls -evalue 1e-10 -outfmt "6 qseqid sseqid qstart qend sstart send length nident pident evalue" Other options: -max_target_seqs:
Review of exercise 1 tblastn -num_threads 2 -db contig -query DH10B.fasta -out blastout.xls -evalue 1e-10 -outfmt "6 qseqid sseqid qstart qend sstart send length nident pident evalue" Other options: -max_target_seqs:
Search for the Dubai in the remap search bar:
 This tutorial is aimed at developing maps for two time periods with in Remap (). In this tutorial we are going to develop a classification water and non-water in Dubai for the year 2000 and the year 2016.
This tutorial is aimed at developing maps for two time periods with in Remap (). In this tutorial we are going to develop a classification water and non-water in Dubai for the year 2000 and the year 2016.
Introduction to Evolutionary Concepts
 Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2009 VMD/MultiSeq
Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2009 VMD/MultiSeq
An Automated Phylogenetic Tree-Based Small Subunit rrna Taxonomy and Alignment Pipeline (STAP)
") An Automated Phylogenetic Tree-Based Small Subunit rrna Taxonomy and Alignment Pipeline (STAP) Dongying Wu 1 *, Amber Hartman 1,6, Naomi Ward 4,5, Jonathan A. Eisen 1,2,3 1 UC Davis Genome Center, University
An Automated Phylogenetic Tree-Based Small Subunit rrna Taxonomy and Alignment Pipeline (STAP) Dongying Wu 1 *, Amber Hartman 1,6, Naomi Ward 4,5, Jonathan A. Eisen 1,2,3 1 UC Davis Genome Center, University
A Browser for Pig Genome Data
 A Browser for Pig Genome Data Thomas Mailund January 2, 2004 This report briefly describe the blast and alignment data available at http://www.daimi.au.dk/ mailund/pig-genome/ hits.html. The report describes
A Browser for Pig Genome Data Thomas Mailund January 2, 2004 This report briefly describe the blast and alignment data available at http://www.daimi.au.dk/ mailund/pig-genome/ hits.html. The report describes
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction. Lesser Tenrec (Echinops telfairi)
") Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction Lesser Tenrec (Echinops telfairi) Goals: 1. Use phylogenetic experimental design theory to select optimal taxa to
Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction Lesser Tenrec (Echinops telfairi) Goals: 1. Use phylogenetic experimental design theory to select optimal taxa to
BIOINFORMATICS LAB AP BIOLOGY
 BIOINFORMATICS LAB AP BIOLOGY Bioinformatics is the science of collecting and analyzing complex biological data. Bioinformatics combines computer science, statistics and biology to allow scientists to
BIOINFORMATICS LAB AP BIOLOGY Bioinformatics is the science of collecting and analyzing complex biological data. Bioinformatics combines computer science, statistics and biology to allow scientists to
Prac%cal Bioinforma%cs for Life Scien%sts. Week 14, Lecture 28. István Albert Bioinforma%cs Consul%ng Center Penn State
 Prac%cal Bioinforma%cs for Life Scien%sts Week 14, Lecture 28 István Albert Bioinforma%cs Consul%ng Center Penn State Final project A group of researchers are interested in studying protein binding loca%ons
Prac%cal Bioinforma%cs for Life Scien%sts Week 14, Lecture 28 István Albert Bioinforma%cs Consul%ng Center Penn State Final project A group of researchers are interested in studying protein binding loca%ons
a-dB. Code assigned:
 This form should be used for all taxonomic proposals. Please complete all those modules that are applicable (and then delete the unwanted sections). For guidance, see the notes written in blue and the
This form should be used for all taxonomic proposals. Please complete all those modules that are applicable (and then delete the unwanted sections). For guidance, see the notes written in blue and the
HOW TO USE MIKANA. 1. Decompress the zip file MATLAB.zip. This will create the directory MIKANA.
 HOW TO USE MIKANA MIKANA (Method to Infer Kinetics And Network Architecture) is a novel computational method to infer reaction mechanisms and estimate the kinetic parameters of biochemical pathways from
HOW TO USE MIKANA MIKANA (Method to Infer Kinetics And Network Architecture) is a novel computational method to infer reaction mechanisms and estimate the kinetic parameters of biochemical pathways from
PNmerger: a Cytoscape plugin to merge biological pathways and protein interaction networks
 PNmerger: a Cytoscape plugin to merge biological pathways and protein interaction networks http://www.hupo.org.cn/pnmerger Fuchu He E-mail: hefc@nic.bmi.ac.cn Tel: 86-10-68171208 FAX: 86-10-68214653 Yunping
PNmerger: a Cytoscape plugin to merge biological pathways and protein interaction networks http://www.hupo.org.cn/pnmerger Fuchu He E-mail: hefc@nic.bmi.ac.cn Tel: 86-10-68171208 FAX: 86-10-68214653 Yunping
E-EROS TUTORIAL Encyclopedia of Reagents for Organic Synthesis at the UIUC Chemistry Library
 E-EROS TUTORIAL Encyclopedia of Reagents for Organic Synthesis at the UIUC Chemistry Library For any questions about e-eros please contact Tina Chrzastowski or call (217) 333-3737. Introduction About e-eros:
E-EROS TUTORIAL Encyclopedia of Reagents for Organic Synthesis at the UIUC Chemistry Library For any questions about e-eros please contact Tina Chrzastowski or call (217) 333-3737. Introduction About e-eros:
Geodatabases and ArcCatalog
 Geodatabases and ArcCatalog Francisco Olivera, Ph.D., P.E. Srikanth Koka Lauren Walker Aishwarya Vijaykumar Keri Clary Department of Civil Engineering April 21, 2014 Contents Geodatabases and ArcCatalog...
Geodatabases and ArcCatalog Francisco Olivera, Ph.D., P.E. Srikanth Koka Lauren Walker Aishwarya Vijaykumar Keri Clary Department of Civil Engineering April 21, 2014 Contents Geodatabases and ArcCatalog...
Comparative Bioinformatics Midterm II Fall 2004
 Comparative Bioinformatics Midterm II Fall 2004 Objective Answer, part I: For each of the following, select the single best answer or completion of the phrase. (3 points each) 1. Deinococcus radiodurans
Comparative Bioinformatics Midterm II Fall 2004 Objective Answer, part I: For each of the following, select the single best answer or completion of the phrase. (3 points each) 1. Deinococcus radiodurans
ProMass Deconvolution User Training. Novatia LLC January, 2013
 ProMass Deconvolution User Training Novatia LLC January, 2013 Overview General info about ProMass Features Basics of how ProMass Deconvolution works Example Spectra Manual Deconvolution with ProMass Deconvolution
ProMass Deconvolution User Training Novatia LLC January, 2013 Overview General info about ProMass Features Basics of how ProMass Deconvolution works Example Spectra Manual Deconvolution with ProMass Deconvolution
DRUG DISCOVERY TODAY ELN ELN. Chemistry. Biology. Known ligands. DBs. Generate chemistry ideas. Check chemical feasibility In-house.
 DRUG DISCOVERY TODAY Known ligands Chemistry ELN DBs Knowledge survey Therapeutic target Generate chemistry ideas Check chemical feasibility In-house Analyze SAR Synthesize or buy Report Test Journals
DRUG DISCOVERY TODAY Known ligands Chemistry ELN DBs Knowledge survey Therapeutic target Generate chemistry ideas Check chemical feasibility In-house Analyze SAR Synthesize or buy Report Test Journals
OECD QSAR Toolbox v.3.3
 OECD QSAR Toolbox v.3.3 Step-by-step example on how to predict the skin sensitisation potential of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific Aims Read
OECD QSAR Toolbox v.3.3 Step-by-step example on how to predict the skin sensitisation potential of a chemical by read-across based on an analogue approach Outlook Background Objectives Specific Aims Read
K-means-based Feature Learning for Protein Sequence Classification
 K-means-based Feature Learning for Protein Sequence Classification Paul Melman and Usman W. Roshan Department of Computer Science, NJIT Newark, NJ, 07102, USA pm462@njit.edu, usman.w.roshan@njit.edu Abstract
K-means-based Feature Learning for Protein Sequence Classification Paul Melman and Usman W. Roshan Department of Computer Science, NJIT Newark, NJ, 07102, USA pm462@njit.edu, usman.w.roshan@njit.edu Abstract
Browsing Genes and Genomes with Ensembl
 Training materials Ensembl training materials are protected by a CC BY license http://creativecommons.org/licenses/by/4.0/ If you wish to re-use these materials, please credit Ensembl for their creation
Training materials Ensembl training materials are protected by a CC BY license http://creativecommons.org/licenses/by/4.0/ If you wish to re-use these materials, please credit Ensembl for their creation
Tutorial: Structural Analysis of a Protein-Protein Complex
 Molecular Modeling Section (MMS) Department of Pharmaceutical and Pharmacological Sciences University of Padova Via Marzolo 5-35131 Padova (IT) @contact: stefano.moro@unipd.it Tutorial: Structural Analysis
Molecular Modeling Section (MMS) Department of Pharmaceutical and Pharmacological Sciences University of Padova Via Marzolo 5-35131 Padova (IT) @contact: stefano.moro@unipd.it Tutorial: Structural Analysis
The Web of Life arxiv:submit/ [q-bio.pe] 11 Mar 2014
![The Web of Life arxiv:submit/ [q-bio.pe] 11 Mar 2014](/thumbs/93/113869982.jpg "The Web of Life arxiv:submit/ [q-bio.pe] 11 Mar 2014") The Web of Life arxiv:submit/0930921 [q-bio.pe] 11 Mar 2014 Miguel A. Fortuna, Raúl Ortega, and Jordi Bascompte Integrative Ecology Group Estación Biológica de Doñana (EBD-CSIC) Seville, Spain To whom
The Web of Life arxiv:submit/0930921 [q-bio.pe] 11 Mar 2014 Miguel A. Fortuna, Raúl Ortega, and Jordi Bascompte Integrative Ecology Group Estación Biológica de Doñana (EBD-CSIC) Seville, Spain To whom