Phylogene)cs. IMBB 2016 BecA- ILRI Hub, Nairobi May 9 20, Joyce Nzioki
|
|
|
- Norah Griffin
- 5 years ago
- Views:
Transcription



1 Phylogene)cs IMBB 2016 BecA- ILRI Hub, Nairobi May 9 20, 2016 Joyce Nzioki
2 Phylogenetics The study of evolutionary relatedness of organisms. Derived from two Greek words:» Phle/Phylon: Tribe/Race» Genetikos: Relative to Birth
3 Phylogene7cs Evolu7on is the change in distribu7on of allele frequencies from one genera7on to the next. Similarity in sequenced data is taken as an indica7on of evolu7onary relatedness. Sequence difference is taken as a measure of evolu7onary divergence. Progression rules: as an organism is more distant from its ancestor their characters are more evolved.
4 Phylogene7cs Phylogeny can be drawn from molecular data (DNA/RNA/Proteins) or morphological data Taxonomy is informed from phylogene7cs Phylogene7c trees are graphical representa7on of the evolu7onary rela7onships through 7me or gene7c distance.
5 Aspects of phylogeny We will look at the major aspects of phylogenies and how they can be interpreted. 1. Topology 2. Branches 3. Nodes 4. Confidence
6 Phylogene7c tree This is a branching diagram that infers evolu7onary rela7onship of various species based on their physical or gene7c traits. Leaves/Operational taxonomic units (OTUs) Branches Internal nodes /Hypothetical taxonomic units (HTUs) Root

7 Topology branching structure of a tree The three top trees have common topologies. A and B are more closely to one another than to C. The bo[om box had different rela7onships

8 Alterna7ve representa7ve of phylogenies The same topology can be drawn in different ways the common format are shown above. Diagonal and rectangular formats commonly used for publica7on Curved format used in review papers to represent summaries of phylogenies Radial format is used to show un- rooted trees Circular format is used to represent large phylogenies.

9 Branches Branches show the path of transmission of gene7c informa7on from one genera7on to the next. Branch lengths indicate gene7c change i.e. the longer the branch, the more gene7c change has occurred. Informa7ve branch lengths are drawn to scale and indicate the number of subs7tu7ons per site A naive method of es7ma7ng the gene7c change is coun7ng the number of differences and dividing by the sequence length i.e. (1/10 = 0.1 ) though this doesn t account for mul7ple subs7tu7ons. To overcome this issues we use evolu7onary models to infer gene7c changes that occur
10 Nodes Tips /OTUs /external nodes the sequences sampled for phylogeny. Internal nodes occur at the point where more that one branch meet and represent the ancestral sequence / hypothe7cal common ancestor. The root represents the most recent common ancestor.
11 Rooted vs Un- rooted tree Un- rooted tree Does not show direc7on of evolu7on / ancestry Rooted tree Direc7on of evolu7on indicated as moving away from the root
12 Roo7ng a tree Two methods are known for tree rou7ng: 1. Outgroup Criteria: include in the analysis a group of sequences known as a priori to be external to the group in study; the root is by necessity the branch joining the outgroup and the other sequences 2. Midpoint roo)ng: this method makes the assump7on that all lineages are supported to have evolved with the same rate since divergence from their common ancestor. The root is at the equidistant point from all tree leaves.
13 Roo7ng a tree with an outgroup This is the use of an organism or group of organisms (outgroup) that are more evolu7onary distant to the group in study (internal group). The common ancestor is therefore placed between the internal group and the outgroup. This effec7vely roots the tree and evolu7onary distances will be rela7ve to this point. (gives a direc7on of evolu7on) Ingroup Outgroup
14 Selec7ng an outgroup An outgroup should not be too distantly related to the internal group, this results in very long branch lengths that distort the remaining branches rendering the topology unreliable. The outgroup should also not be too closely related to the internal group this may not make a true outgroup. Using various outgroup species may be[er balance the final tree branching.
15 Confidence Inferring phylogenies is inherently an uncertain process. Approaches used to es7mate our confidence in the inferred tree topology: bootstraps, likelihood and Bayesian approaches. This course we will focus on bootstrap. Typically these are shown on the phylogeny. Indica7ng the percentage confidence of a branch. Interpre7ng the exact meaning of confidence values is s7ll an area of debate but generally >50% is acceptable provided an appropriate evolu7onary model was used.
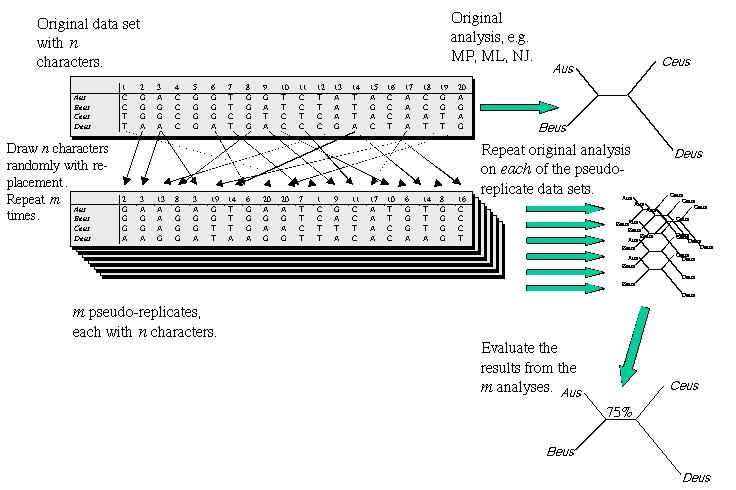
16 Bootstrapping Bootstrapping is commonly used test of reliability of inferred phylogene7c tree. A single tree may not be credible given the dependencies involved: (characters, evolu7onary model, parameters). Bootstrapping is done by genera7ng replicas of your data (arrange character posi7ons at random, to create a series of bootstrap samples of same size as original data) The bootstrap datasets are analyzed looking for consistency. Varia7on among the datasets is used to es7mate error involved in making es7mates in the original data
17
. Fish are equally related to mice as they are to frogs.")
18 Interpre7ng pa[erns of relatedness Humans are more closely related to mice than lizard (share a more recent common ancestor). Frogs are more closely related to lizards than they are to fish (as they share a common ancestor with lizard more recent than fish). Fish are equally related to mice as they are to frogs. Mice and frogs both share the same common ancestor (black spot ) with fish. If you rotate the branches to change topology, the biological meaning remains.


19 Major stages in phylogene7c analysis
 are")
20 Building a phylogene7c tree Staring point :a set of homologous aligned DNA or protein sequences Quality of the alignment is essen7al, unreliable parts of the alignment are omi[ed Most methods only take into account subs7tu7ons gaps(dele7ons/inser7ons) are not used.
21
22 The gene compared must evolve at a rate comparable to the divergence 7me of the organism; for example: 18S rrna gene for phylum- level divergences since it evolved slowly Hemoglobin genes for mammalian orders. Mitochondrial DNA for species divergences within a genus Repe77ve DNA sequences (e.g. microsatellites) for individuals within a species
23 Method of building a tree 1. Distance methods 2. Character based methods 1. Maximum parsimony 2. Maximum likelihood 3. Bayesian inference
24 Distance methods Start from a mul7ple sequence alignment Make a matrix of pairwise distances Build a phylogene7c tree. E.g. Neighbor joining and UPGMA trees
25 Distance methods To es7mate evolu7onary distances between sequences there is need for sta7s7cal / evolu7onary models. Sta7s7cal models es7mate for evolu7onary distance while accoun7ng for residue subs7tu7on and homoplasy. Juke- Cantors: good for distances <10% Kimura- 2: distance 10-30% and transi7ons ~= transversions Tamura: distances 10-30% and strong G+C bias Jin- Nei γ: distance 10-30% and varying transi7on- transver7on rates Tajima- Nei: distances % These evolu7onary distances are then converted into a distance matrix used in building the tree
26 Character based methods This analyses any set of discrete character, that is each posi7on in an aligned sequence character. All character can be analyzed separately and independently of one another. These include: 1. Maximum Parsimony (MP) 2. Maximum Likelihood (ML) 3. Bayesian methods
27 Maximum Parsimony Parsimony involves evalua7ng all possible trees and giving each a score based on the number of evolu7onary changes that are needed to explain the observed data. The best tree is the one that requires the fewest base changes for all sequences to derive from a common ancestor
28 Maximum Likelihood Maximum Likelihood evaluates the topologies of different trees given a par7cular evolu7on model and picks the best one according to the likelihood score. (tree with the highest likelihood) It considers all characters and looks for trees that best suit a given evolu7on model. It is possibly more accurate than Maximum parsimony if the appropriate model is chosen.
29 summary UPGMA assumes molecular clock, so provides a rooted tree (this assump7on may be too strong in some cases) Neighbor joining has been proved to create correct trees when evolu7onary rates vary. Maximum Parsimony is good for closely related sequences Maximum likelihood methods is the general of all three.
on phylogene7cs gives accurate pa[erns of relatedness that now inform the Linnaen")

30 Applications of Phylogenetics 1. Classifica)on phylogene7cs gives accurate pa[erns of relatedness that now inform the Linnaen classifica7on of new species. 2. Forensics - Access DNA evidence presented in court. 3. Origin of pathogens learn about new pathogen outbreak and source of transmission. 4. Conserva)on - can inform conserva7on policies of species becoming ex7nct.


31
32 Thanks Thanks to Anne Jores and EBI online training for the some of the slides presented
POPULATION GENETICS Winter 2005 Lecture 17 Molecular phylogenetics
 POPULATION GENETICS Winter 2005 Lecture 17 Molecular phylogenetics - in deriving a phylogeny our goal is simply to reconstruct the historical relationships between a group of taxa. - before we review the
POPULATION GENETICS Winter 2005 Lecture 17 Molecular phylogenetics - in deriving a phylogeny our goal is simply to reconstruct the historical relationships between a group of taxa. - before we review the
Dr. Amira A. AL-Hosary
 Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut
 Faculty of Veterinary Medicine Assiut") Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
"Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky
 MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
BINF6201/8201. Molecular phylogenetic methods
 BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
Phylogenetics: Building Phylogenetic Trees
 1 Phylogenetics: Building Phylogenetic Trees COMP 571 Luay Nakhleh, Rice University 2 Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary model should
1 Phylogenetics: Building Phylogenetic Trees COMP 571 Luay Nakhleh, Rice University 2 Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary model should
Constructing Evolutionary/Phylogenetic Trees
 Constructing Evolutionary/Phylogenetic Trees 2 broad categories: istance-based methods Ultrametric Additive: UPGMA Transformed istance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
Constructing Evolutionary/Phylogenetic Trees 2 broad categories: istance-based methods Ultrametric Additive: UPGMA Transformed istance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
Phylogenetic inference
 Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
What is Phylogenetics
 What is Phylogenetics Phylogenetics is the area of research concerned with finding the genetic connections and relationships between species. The basic idea is to compare specific characters (features)
What is Phylogenetics Phylogenetics is the area of research concerned with finding the genetic connections and relationships between species. The basic idea is to compare specific characters (features)
Phylogenetics: Building Phylogenetic Trees. COMP Fall 2010 Luay Nakhleh, Rice University
 Phylogenetics: Building Phylogenetic Trees COMP 571 - Fall 2010 Luay Nakhleh, Rice University Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary
Phylogenetics: Building Phylogenetic Trees COMP 571 - Fall 2010 Luay Nakhleh, Rice University Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary
CSCI1950 Z Computa3onal Methods for Biology* (*Working Title) Lecture 1. Ben Raphael January 21, Course Par3culars
 Lecture 1. Ben Raphael January 21, Course Par3culars") CSCI1950 Z Computa3onal Methods for Biology* (*Working Title) Lecture 1 Ben Raphael January 21, 2009 Course Par3culars Three major topics 1. Phylogeny: ~50% lectures 2. Func3onal Genomics: ~25% lectures
CSCI1950 Z Computa3onal Methods for Biology* (*Working Title) Lecture 1 Ben Raphael January 21, 2009 Course Par3culars Three major topics 1. Phylogeny: ~50% lectures 2. Func3onal Genomics: ~25% lectures
Phylogeny Fig Overview: Inves8ga8ng the Tree of Life Phylogeny Systema8cs
 Ch. 26 Phylogeny BIOL 22 Fig. Fig.26 26 Overview: Inves8ga8ng the Tree of Life Phylogeny evoluonary history of a species or group of related species Systema8cs classifies organisms and determines their
Ch. 26 Phylogeny BIOL 22 Fig. Fig.26 26 Overview: Inves8ga8ng the Tree of Life Phylogeny evoluonary history of a species or group of related species Systema8cs classifies organisms and determines their
C3020 Molecular Evolution. Exercises #3: Phylogenetics
 C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
Phylogenetic Tree Reconstruction
 I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
Multiple Sequence Alignment. Sequences
 Multiple Sequence Alignment Sequences > YOR020c mstllksaksivplmdrvlvqrikaqaktasglylpe knveklnqaevvavgpgftdangnkvvpqvkvgdqvl ipqfggstiklgnddevilfrdaeilakiakd > crassa mattvrsvksliplldrvlvqrvkaeaktasgiflpe
Multiple Sequence Alignment Sequences > YOR020c mstllksaksivplmdrvlvqrikaqaktasglylpe knveklnqaevvavgpgftdangnkvvpqvkvgdqvl ipqfggstiklgnddevilfrdaeilakiakd > crassa mattvrsvksliplldrvlvqrvkaeaktasgiflpe
A (short) introduction to phylogenetics
 introduction to phylogenetics") A (short) introduction to phylogenetics Thibaut Jombart, Marie-Pauline Beugin MRC Centre for Outbreak Analysis and Modelling Imperial College London Genetic data analysis with PR Statistics, Millport Field
A (short) introduction to phylogenetics Thibaut Jombart, Marie-Pauline Beugin MRC Centre for Outbreak Analysis and Modelling Imperial College London Genetic data analysis with PR Statistics, Millport Field
UoN, CAS, DBSC BIOL102 lecture notes by: Dr. Mustafa A. Mansi. The Phylogenetic Systematics (Phylogeny and Systematics)
") - Phylogeny? - Systematics? The Phylogenetic Systematics (Phylogeny and Systematics) - Phylogenetic systematics? Connection between phylogeny and classification. - Phylogenetic systematics informs the
- Phylogeny? - Systematics? The Phylogenetic Systematics (Phylogeny and Systematics) - Phylogenetic systematics? Connection between phylogeny and classification. - Phylogenetic systematics informs the
Classification and Phylogeny
 Classification and Phylogeny The diversity of life is great. To communicate about it, there must be a scheme for organization. There are many species that would be difficult to organize without a scheme
Classification and Phylogeny The diversity of life is great. To communicate about it, there must be a scheme for organization. There are many species that would be difficult to organize without a scheme
Mul$ple Sequence Alignment Methods. Tandy Warnow Departments of Bioengineering and Computer Science h?p://tandy.cs.illinois.edu
 Mul$ple Sequence Alignment Methods Tandy Warnow Departments of Bioengineering and Computer Science h?p://tandy.cs.illinois.edu Species Tree Orangutan Gorilla Chimpanzee Human From the Tree of the Life
Mul$ple Sequence Alignment Methods Tandy Warnow Departments of Bioengineering and Computer Science h?p://tandy.cs.illinois.edu Species Tree Orangutan Gorilla Chimpanzee Human From the Tree of the Life
Ch. 26 Phylogeny BIOL 221
 Ch. 26 Phylogeny BIOL 22 Fig. 26- Phylogeny Overview: Inves8ga8ng the Tree of Life evoluonary history of a species Systema8cs or group of related species classifies organisms and determines their evoluonary
Ch. 26 Phylogeny BIOL 22 Fig. 26- Phylogeny Overview: Inves8ga8ng the Tree of Life evoluonary history of a species Systema8cs or group of related species classifies organisms and determines their evoluonary
Classification and Phylogeny
 Classification and Phylogeny The diversity it of life is great. To communicate about it, there must be a scheme for organization. There are many species that would be difficult to organize without a scheme
Classification and Phylogeny The diversity it of life is great. To communicate about it, there must be a scheme for organization. There are many species that would be difficult to organize without a scheme
How to read and make phylogenetic trees Zuzana Starostová
 How to read and make phylogenetic trees Zuzana Starostová How to make phylogenetic trees? Workflow: obtain DNA sequence quality check sequence alignment calculating genetic distances phylogeny estimation
How to read and make phylogenetic trees Zuzana Starostová How to make phylogenetic trees? Workflow: obtain DNA sequence quality check sequence alignment calculating genetic distances phylogeny estimation
Phylogenetic analyses. Kirsi Kostamo
 Phylogenetic analyses Kirsi Kostamo The aim: To construct a visual representation (a tree) to describe the assumed evolution occurring between and among different groups (individuals, populations, species,
Phylogenetic analyses Kirsi Kostamo The aim: To construct a visual representation (a tree) to describe the assumed evolution occurring between and among different groups (individuals, populations, species,
Constructing Evolutionary/Phylogenetic Trees
 Constructing Evolutionary/Phylogenetic Trees 2 broad categories: Distance-based methods Ultrametric Additive: UPGMA Transformed Distance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
Constructing Evolutionary/Phylogenetic Trees 2 broad categories: Distance-based methods Ultrametric Additive: UPGMA Transformed Distance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
 Tree of Life iological Sequence nalysis Chapter http://tolweb.org/tree/ Phylogenetic Prediction ll organisms on Earth have a common ancestor. ll species are related. The relationship is called a phylogeny
Tree of Life iological Sequence nalysis Chapter http://tolweb.org/tree/ Phylogenetic Prediction ll organisms on Earth have a common ancestor. ll species are related. The relationship is called a phylogeny
Lecture 6 Phylogenetic Inference
 Lecture 6 Phylogenetic Inference From Darwin s notebook in 1837 Charles Darwin Willi Hennig From The Origin in 1859 Cladistics Phylogenetic inference Willi Hennig, Cladistics 1. Clade, Monophyletic group,
Lecture 6 Phylogenetic Inference From Darwin s notebook in 1837 Charles Darwin Willi Hennig From The Origin in 1859 Cladistics Phylogenetic inference Willi Hennig, Cladistics 1. Clade, Monophyletic group,
Basics on bioinforma-cs Lecture 7. Nunzio D Agostino
 Basics on bioinforma-cs Lecture 7 Nunzio D Agostino nunzio.dagostino@entecra.it; nunzio.dagostino@gmail.com Multiple alignments One sequence plays coy a pair of homologous sequence whisper many aligned
Basics on bioinforma-cs Lecture 7 Nunzio D Agostino nunzio.dagostino@entecra.it; nunzio.dagostino@gmail.com Multiple alignments One sequence plays coy a pair of homologous sequence whisper many aligned
8/23/2014. Phylogeny and the Tree of Life
 Phylogeny and the Tree of Life Chapter 26 Objectives Explain the following characteristics of the Linnaean system of classification: a. binomial nomenclature b. hierarchical classification List the major
Phylogeny and the Tree of Life Chapter 26 Objectives Explain the following characteristics of the Linnaean system of classification: a. binomial nomenclature b. hierarchical classification List the major
Chapter 26: Phylogeny and the Tree of Life Phylogenies Show Evolutionary Relationships
 Chapter 26: Phylogeny and the Tree of Life You Must Know The taxonomic categories and how they indicate relatedness. How systematics is used to develop phylogenetic trees. How to construct a phylogenetic
Chapter 26: Phylogeny and the Tree of Life You Must Know The taxonomic categories and how they indicate relatedness. How systematics is used to develop phylogenetic trees. How to construct a phylogenetic
Inferring phylogeny. Constructing phylogenetic trees. Tõnu Margus. Bioinformatics MTAT
 Inferring phylogeny Constructing phylogenetic trees Tõnu Margus Contents What is phylogeny? How/why it is possible to infer it? Representing evolutionary relationships on trees What type questions questions
Inferring phylogeny Constructing phylogenetic trees Tõnu Margus Contents What is phylogeny? How/why it is possible to infer it? Representing evolutionary relationships on trees What type questions questions
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Distance Methods Character Methods
Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Distance Methods Character Methods
Phylogenetic Trees. What They Are Why We Do It & How To Do It. Presented by Amy Harris Dr Brad Morantz
 Phylogenetic Trees What They Are Why We Do It & How To Do It Presented by Amy Harris Dr Brad Morantz Overview What is a phylogenetic tree Why do we do it How do we do it Methods and programs Parallels
Phylogenetic Trees What They Are Why We Do It & How To Do It Presented by Amy Harris Dr Brad Morantz Overview What is a phylogenetic tree Why do we do it How do we do it Methods and programs Parallels
Molecular Phylogenetics (part 1 of 2) Computational Biology Course João André Carriço
 Computational Biology Course João André Carriço") Molecular Phylogenetics (part 1 of 2) Computational Biology Course João André Carriço jcarrico@fm.ul.pt Charles Darwin (1809-1882) Charles Darwin s tree of life in Notebook B, 1837-1838 Ernst Haeckel (1934-1919)
Molecular Phylogenetics (part 1 of 2) Computational Biology Course João André Carriço jcarrico@fm.ul.pt Charles Darwin (1809-1882) Charles Darwin s tree of life in Notebook B, 1837-1838 Ernst Haeckel (1934-1919)
Phylogenetic Analysis. Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center
 Phylogenetic Analysis Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center Outline Basic Concepts Tree Construction Methods Distance-based methods
Phylogenetic Analysis Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center Outline Basic Concepts Tree Construction Methods Distance-based methods
Phylogenetic Trees. Phylogenetic Trees Five. Phylogeny: Inference Tool. Phylogeny Terminology. Picture of Last Quagga. Importance of Phylogeny 5.
 Five Sami Khuri Department of Computer Science San José State University San José, California, USA sami.khuri@sjsu.edu v Distance Methods v Character Methods v Molecular Clock v UPGMA v Maximum Parsimony
Five Sami Khuri Department of Computer Science San José State University San José, California, USA sami.khuri@sjsu.edu v Distance Methods v Character Methods v Molecular Clock v UPGMA v Maximum Parsimony
Phylogenetic Analysis
 Phylogenetic Analysis Aristotle Through classification, one might discover the essence and purpose of species. Nelson & Platnick (1981) Systematics and Biogeography Carl Linnaeus Swedish botanist (1700s)
Phylogenetic Analysis Aristotle Through classification, one might discover the essence and purpose of species. Nelson & Platnick (1981) Systematics and Biogeography Carl Linnaeus Swedish botanist (1700s)
Phylogenetic Analysis
 Phylogenetic Analysis Aristotle Through classification, one might discover the essence and purpose of species. Nelson & Platnick (1981) Systematics and Biogeography Carl Linnaeus Swedish botanist (1700s)
Phylogenetic Analysis Aristotle Through classification, one might discover the essence and purpose of species. Nelson & Platnick (1981) Systematics and Biogeography Carl Linnaeus Swedish botanist (1700s)
Phylogenetic Analysis
 Phylogenetic Analysis Aristotle Through classification, one might discover the essence and purpose of species. Nelson & Platnick (1981) Systematics and Biogeography Carl Linnaeus Swedish botanist (1700s)
Phylogenetic Analysis Aristotle Through classification, one might discover the essence and purpose of species. Nelson & Platnick (1981) Systematics and Biogeography Carl Linnaeus Swedish botanist (1700s)
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Phylogeny 9/8/2014. Evolutionary Relationships. Data Supporting Phylogeny. Chapter 26
 Phylogeny Chapter 26 Taxonomy Taxonomy: ordered division of organisms into categories based on a set of characteristics used to assess similarities and differences Carolus Linnaeus developed binomial nomenclature,
Phylogeny Chapter 26 Taxonomy Taxonomy: ordered division of organisms into categories based on a set of characteristics used to assess similarities and differences Carolus Linnaeus developed binomial nomenclature,
Phylogeny. November 7, 2017
 Phylogeny November 7, 2017 Phylogenetics Phylon = tribe/race, genetikos = relative to birth Phylogenetics: study of evolutionary relationships among organisms, sequences, or anything in between Related
Phylogeny November 7, 2017 Phylogenetics Phylon = tribe/race, genetikos = relative to birth Phylogenetics: study of evolutionary relationships among organisms, sequences, or anything in between Related
Chapter 19: Taxonomy, Systematics, and Phylogeny
 Chapter 19: Taxonomy, Systematics, and Phylogeny AP Curriculum Alignment Chapter 19 expands on the topics of phylogenies and cladograms, which are important to Big Idea 1. In order for students to understand
Chapter 19: Taxonomy, Systematics, and Phylogeny AP Curriculum Alignment Chapter 19 expands on the topics of phylogenies and cladograms, which are important to Big Idea 1. In order for students to understand
Biology 211 (2) Week 1 KEY!
 Week 1 KEY!") Biology 211 (2) Week 1 KEY Chapter 1 KEY FIGURES: 1.2, 1.3, 1.4, 1.5, 1.6, 1.7 VOCABULARY: Adaptation: a trait that increases the fitness Cells: a developed, system bound with a thin outer layer made of
Biology 211 (2) Week 1 KEY Chapter 1 KEY FIGURES: 1.2, 1.3, 1.4, 1.5, 1.6, 1.7 VOCABULARY: Adaptation: a trait that increases the fitness Cells: a developed, system bound with a thin outer layer made of
Classifica=on of Organisms. Chapter 7. A Taxonomic Hierarchy 8/31/18. Systema=cs or taxonomy. Animal Taxonomy, Phylogeny, and Organiza=on.
 Classifica=on of Organisms Chapter 7 Systema=cs or taxonomy Study of the kinds and diversity of organisms and of the evolu=onary rela=onships among them Animal Taxonomy, Phylogeny, and Organiza=on A Taxonomic
Classifica=on of Organisms Chapter 7 Systema=cs or taxonomy Study of the kinds and diversity of organisms and of the evolu=onary rela=onships among them Animal Taxonomy, Phylogeny, and Organiza=on A Taxonomic
Macroevolution Part I: Phylogenies
 Macroevolution Part I: Phylogenies Taxonomy Classification originated with Carolus Linnaeus in the 18 th century. Based on structural (outward and inward) similarities Hierarchal scheme, the largest most
Macroevolution Part I: Phylogenies Taxonomy Classification originated with Carolus Linnaeus in the 18 th century. Based on structural (outward and inward) similarities Hierarchal scheme, the largest most
Reconstructing the history of lineages
 Reconstructing the history of lineages Class outline Systematics Phylogenetic systematics Phylogenetic trees and maps Class outline Definitions Systematics Phylogenetic systematics/cladistics Systematics
Reconstructing the history of lineages Class outline Systematics Phylogenetic systematics Phylogenetic trees and maps Class outline Definitions Systematics Phylogenetic systematics/cladistics Systematics
Anatomy of a tree. clade is group of organisms with a shared ancestor. a monophyletic group shares a single common ancestor = tapirs-rhinos-horses
 Anatomy of a tree outgroup: an early branching relative of the interest groups sister taxa: taxa derived from the same recent ancestor polytomy: >2 taxa emerge from a node Anatomy of a tree clade is group
Anatomy of a tree outgroup: an early branching relative of the interest groups sister taxa: taxa derived from the same recent ancestor polytomy: >2 taxa emerge from a node Anatomy of a tree clade is group
Introduction to characters and parsimony analysis
 Introduction to characters and parsimony analysis Genetic Relationships Genetic relationships exist between individuals within populations These include ancestordescendent relationships and more indirect
Introduction to characters and parsimony analysis Genetic Relationships Genetic relationships exist between individuals within populations These include ancestordescendent relationships and more indirect
Cladistics and Bioinformatics Questions 2013
 AP Biology Name Cladistics and Bioinformatics Questions 2013 1. The following table shows the percentage similarity in sequences of nucleotides from a homologous gene derived from five different species
AP Biology Name Cladistics and Bioinformatics Questions 2013 1. The following table shows the percentage similarity in sequences of nucleotides from a homologous gene derived from five different species
C.DARWIN ( )
") C.DARWIN (1809-1882) LAMARCK Each evolutionary lineage has evolved, transforming itself, from a ancestor appeared by spontaneous generation DARWIN All organisms are historically interconnected. Their relationships
C.DARWIN (1809-1882) LAMARCK Each evolutionary lineage has evolved, transforming itself, from a ancestor appeared by spontaneous generation DARWIN All organisms are historically interconnected. Their relationships
Bioinformatics tools for phylogeny and visualization. Yanbin Yin
 Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
Phylogenetics. Applications of phylogenetics. Unrooted networks vs. rooted trees. Outline
 Phylogenetics Todd Vision iology 522 March 26, 2007 pplications of phylogenetics Studying organismal or biogeographic history Systematics ating events in the fossil record onservation biology Studying
Phylogenetics Todd Vision iology 522 March 26, 2007 pplications of phylogenetics Studying organismal or biogeographic history Systematics ating events in the fossil record onservation biology Studying
9/30/11. Evolution theory. Phylogenetic Tree Reconstruction. Phylogenetic trees (binary trees) Phylogeny (phylogenetic tree)
 Phylogeny (phylogenetic tree)") I9 Introduction to Bioinformatics, 0 Phylogenetic ree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & omputing, IUB Evolution theory Speciation Evolution of new organisms is driven by
I9 Introduction to Bioinformatics, 0 Phylogenetic ree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & omputing, IUB Evolution theory Speciation Evolution of new organisms is driven by
Classification, Phylogeny yand Evolutionary History
 Classification, Phylogeny yand Evolutionary History The diversity of life is great. To communicate about it, there must be a scheme for organization. There are many species that would be difficult to organize
Classification, Phylogeny yand Evolutionary History The diversity of life is great. To communicate about it, there must be a scheme for organization. There are many species that would be difficult to organize
Bioinformatics 1. Sepp Hochreiter. Biology, Sequences, Phylogenetics Part 4. Bioinformatics 1: Biology, Sequences, Phylogenetics
 Bioinformatics 1 Biology, Sequences, Phylogenetics Part 4 Sepp Hochreiter Klausur Mo. 30.01.2011 Zeit: 15:30 17:00 Raum: HS14 Anmeldung Kusss Contents Methods and Bootstrapping of Maximum Methods Methods
Bioinformatics 1 Biology, Sequences, Phylogenetics Part 4 Sepp Hochreiter Klausur Mo. 30.01.2011 Zeit: 15:30 17:00 Raum: HS14 Anmeldung Kusss Contents Methods and Bootstrapping of Maximum Methods Methods
Phylogenetics. BIOL 7711 Computational Bioscience
 Consortium for Comparative Genomics! University of Colorado School of Medicine Phylogenetics BIOL 7711 Computational Bioscience Biochemistry and Molecular Genetics Computational Bioscience Program Consortium
Consortium for Comparative Genomics! University of Colorado School of Medicine Phylogenetics BIOL 7711 Computational Bioscience Biochemistry and Molecular Genetics Computational Bioscience Program Consortium
CSCI1950 Z Computa4onal Methods for Biology Lecture 5
 CSCI1950 Z Computa4onal Methods for Biology Lecture 5 Ben Raphael February 6, 2009 hip://cs.brown.edu/courses/csci1950 z/ Alignment vs. Distance Matrix Mouse: ACAGTGACGCCACACACGT Gorilla: CCTGCGACGTAACAAACGC
CSCI1950 Z Computa4onal Methods for Biology Lecture 5 Ben Raphael February 6, 2009 hip://cs.brown.edu/courses/csci1950 z/ Alignment vs. Distance Matrix Mouse: ACAGTGACGCCACACACGT Gorilla: CCTGCGACGTAACAAACGC
Chapter 26 Phylogeny and the Tree of Life
 Chapter 26 Phylogeny and the Tree of Life Biologists estimate that there are about 5 to 100 million species of organisms living on Earth today. Evidence from morphological, biochemical, and gene sequence
Chapter 26 Phylogeny and the Tree of Life Biologists estimate that there are about 5 to 100 million species of organisms living on Earth today. Evidence from morphological, biochemical, and gene sequence
PHYLOGENY & THE TREE OF LIFE
 PHYLOGENY & THE TREE OF LIFE PREFACE In this powerpoint we learn how biologists distinguish and categorize the millions of species on earth. Early we looked at the process of evolution here we look at
PHYLOGENY & THE TREE OF LIFE PREFACE In this powerpoint we learn how biologists distinguish and categorize the millions of species on earth. Early we looked at the process of evolution here we look at
Theory of Evolution Charles Darwin
 Theory of Evolution Charles arwin 858-59: Origin of Species 5 year voyage of H.M.S. eagle (83-36) Populations have variations. Natural Selection & Survival of the fittest: nature selects best adapted varieties
Theory of Evolution Charles arwin 858-59: Origin of Species 5 year voyage of H.M.S. eagle (83-36) Populations have variations. Natural Selection & Survival of the fittest: nature selects best adapted varieties
CSCI1950 Z Computa4onal Methods for Biology Lecture 4. Ben Raphael February 2, hhp://cs.brown.edu/courses/csci1950 z/ Algorithm Summary
 CSCI1950 Z Computa4onal Methods for Biology Lecture 4 Ben Raphael February 2, 2009 hhp://cs.brown.edu/courses/csci1950 z/ Algorithm Summary Parsimony Probabilis4c Method Input Output Sankoff s & Fitch
CSCI1950 Z Computa4onal Methods for Biology Lecture 4 Ben Raphael February 2, 2009 hhp://cs.brown.edu/courses/csci1950 z/ Algorithm Summary Parsimony Probabilis4c Method Input Output Sankoff s & Fitch
Lecture V Phylogeny and Systematics Dr. Kopeny
 Delivered 1/30 and 2/1 Lecture V Phylogeny and Systematics Dr. Kopeny Lecture V How to Determine Evolutionary Relationships: Concepts in Phylogeny and Systematics Textbook Reading: pp 425-433, 435-437
Delivered 1/30 and 2/1 Lecture V Phylogeny and Systematics Dr. Kopeny Lecture V How to Determine Evolutionary Relationships: Concepts in Phylogeny and Systematics Textbook Reading: pp 425-433, 435-437
Phylogeny and the Tree of Life
 LECTURE PRESENTATIONS For CAMPBELL BIOLOGY, NINTH EDITION Jane B. Reece, Lisa A. Urry, Michael L. Cain, Steven A. Wasserman, Peter V. Minorsky, Robert B. Jackson Chapter 26 Phylogeny and the Tree of Life
LECTURE PRESENTATIONS For CAMPBELL BIOLOGY, NINTH EDITION Jane B. Reece, Lisa A. Urry, Michael L. Cain, Steven A. Wasserman, Peter V. Minorsky, Robert B. Jackson Chapter 26 Phylogeny and the Tree of Life
Phylogeny and Evolution. Gina Cannarozzi ETH Zurich Institute of Computational Science
 Phylogeny and Evolution Gina Cannarozzi ETH Zurich Institute of Computational Science History Aristotle (384-322 BC) classified animals. He found that dolphins do not belong to the fish but to the mammals.
Phylogeny and Evolution Gina Cannarozzi ETH Zurich Institute of Computational Science History Aristotle (384-322 BC) classified animals. He found that dolphins do not belong to the fish but to the mammals.
Molecular phylogeny - Using molecular sequences to infer evolutionary relationships. Tore Samuelsson Feb 2016
 Molecular phylogeny - Using molecular sequences to infer evolutionary relationships Tore Samuelsson Feb 2016 Molecular phylogeny is being used in the identification and characterization of new pathogens,
Molecular phylogeny - Using molecular sequences to infer evolutionary relationships Tore Samuelsson Feb 2016 Molecular phylogeny is being used in the identification and characterization of new pathogens,
Phylogeny: building the tree of life
 Phylogeny: building the tree of life Dr. Fayyaz ul Amir Afsar Minhas Department of Computer and Information Sciences Pakistan Institute of Engineering & Applied Sciences PO Nilore, Islamabad, Pakistan
Phylogeny: building the tree of life Dr. Fayyaz ul Amir Afsar Minhas Department of Computer and Information Sciences Pakistan Institute of Engineering & Applied Sciences PO Nilore, Islamabad, Pakistan
Sec$on 9. Evolu$onary Rela$onships
 Sec$on 9 Evolu$onary Rela$onships Sec$on 9 Learning Goals Explain why the ribosomal 16S gene is a good marker for molecular phylogene$c comparisons. Be able to interpret a phylogene$c tree. Explain the
Sec$on 9 Evolu$onary Rela$onships Sec$on 9 Learning Goals Explain why the ribosomal 16S gene is a good marker for molecular phylogene$c comparisons. Be able to interpret a phylogene$c tree. Explain the
Phylogenies & Classifying species (AKA Cladistics & Taxonomy) What are phylogenies & cladograms? How do we read them? How do we estimate them?
 What are phylogenies & cladograms? How do we read them? How do we estimate them?") Phylogenies & Classifying species (AKA Cladistics & Taxonomy) What are phylogenies & cladograms? How do we read them? How do we estimate them? Carolus Linneaus:Systema Naturae (1735) Swedish botanist &
Phylogenies & Classifying species (AKA Cladistics & Taxonomy) What are phylogenies & cladograms? How do we read them? How do we estimate them? Carolus Linneaus:Systema Naturae (1735) Swedish botanist &
Molecular phylogeny How to infer phylogenetic trees using molecular sequences
 Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 2009 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 2009 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Molecular phylogeny How to infer phylogenetic trees using molecular sequences
 Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 200 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 200 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Lecture 11 Friday, October 21, 2011
 Lecture 11 Friday, October 21, 2011 Phylogenetic tree (phylogeny) Darwin and classification: In the Origin, Darwin said that descent from a common ancestral species could explain why the Linnaean system
Lecture 11 Friday, October 21, 2011 Phylogenetic tree (phylogeny) Darwin and classification: In the Origin, Darwin said that descent from a common ancestral species could explain why the Linnaean system
How should we organize the diversity of animal life?
 How should we organize the diversity of animal life? The difference between Taxonomy Linneaus, and Cladistics Darwin What are phylogenies? How do we read them? How do we estimate them? Classification (Taxonomy)
How should we organize the diversity of animal life? The difference between Taxonomy Linneaus, and Cladistics Darwin What are phylogenies? How do we read them? How do we estimate them? Classification (Taxonomy)
The practice of naming and classifying organisms is called taxonomy.
 Chapter 18 Key Idea: Biologists use taxonomic systems to organize their knowledge of organisms. These systems attempt to provide consistent ways to name and categorize organisms. The practice of naming
Chapter 18 Key Idea: Biologists use taxonomic systems to organize their knowledge of organisms. These systems attempt to provide consistent ways to name and categorize organisms. The practice of naming
Lab 06 Phylogenetics, part 1
 Lab 06 Phylogenetics, part 1 phylogeny is a visual representation of a hypothesis about the relationships among a set of organisms. Phylogenetics is the study of phylogenies and and their development.
Lab 06 Phylogenetics, part 1 phylogeny is a visual representation of a hypothesis about the relationships among a set of organisms. Phylogenetics is the study of phylogenies and and their development.
ELE4120 Bioinformatics Tutorial 8
 ELE4120 ioinformatics Tutorial 8 ontent lassifying Organisms Systematics and Speciation Taxonomy and phylogenetics Phenetics versus cladistics Phylogenetic trees iological classification Goal: To develop
ELE4120 ioinformatics Tutorial 8 ontent lassifying Organisms Systematics and Speciation Taxonomy and phylogenetics Phenetics versus cladistics Phylogenetic trees iological classification Goal: To develop
Chapter 26 Phylogeny and the Tree of Life
 Chapter 26 Phylogeny and the Tree of Life Chapter focus Shifting from the process of how evolution works to the pattern evolution produces over time. Phylogeny Phylon = tribe, geny = genesis or origin
Chapter 26 Phylogeny and the Tree of Life Chapter focus Shifting from the process of how evolution works to the pattern evolution produces over time. Phylogeny Phylon = tribe, geny = genesis or origin
Phylogeny and the Tree of Life
 Chapter 26 Phylogeny and the Tree of Life PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from
Chapter 26 Phylogeny and the Tree of Life PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from
Some of these slides have been borrowed from Dr. Paul Lewis, Dr. Joe Felsenstein. Thanks!
 Some of these slides have been borrowed from Dr. Paul Lewis, Dr. Joe Felsenstein. Thanks! Paul has many great tools for teaching phylogenetics at his web site: http://hydrodictyon.eeb.uconn.edu/people/plewis
Some of these slides have been borrowed from Dr. Paul Lewis, Dr. Joe Felsenstein. Thanks! Paul has many great tools for teaching phylogenetics at his web site: http://hydrodictyon.eeb.uconn.edu/people/plewis
Letter to the Editor. Department of Biology, Arizona State University
 Letter to the Editor Traditional Phylogenetic Reconstruction Methods Reconstruct Shallow and Deep Evolutionary Relationships Equally Well Michael S. Rosenberg and Sudhir Kumar Department of Biology, Arizona
Letter to the Editor Traditional Phylogenetic Reconstruction Methods Reconstruct Shallow and Deep Evolutionary Relationships Equally Well Michael S. Rosenberg and Sudhir Kumar Department of Biology, Arizona
Chapter 26. Phylogeny and the Tree of Life. Lecture Presentations by Nicole Tunbridge and Kathleen Fitzpatrick Pearson Education, Inc.
 Chapter 26 Phylogeny and the Tree of Life Lecture Presentations by Nicole Tunbridge and Kathleen Fitzpatrick Investigating the Tree of Life Phylogeny is the evolutionary history of a species or group of
Chapter 26 Phylogeny and the Tree of Life Lecture Presentations by Nicole Tunbridge and Kathleen Fitzpatrick Investigating the Tree of Life Phylogeny is the evolutionary history of a species or group of
PHYLOGENY WHAT IS EVOLUTION? 1/22/2018. Change must occur in a population via allele
 PHYLOGENY EXERCISE 1 AND 2 WHAT IS EVOLUTION? The theory that all living organisms on earth are related and have a common ancestor. These organism have changed over time and are continuing to change. Changes
PHYLOGENY EXERCISE 1 AND 2 WHAT IS EVOLUTION? The theory that all living organisms on earth are related and have a common ancestor. These organism have changed over time and are continuing to change. Changes
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Outline. Classification of Living Things
 Outline Classification of Living Things Chapter 20 Mader: Biology 8th Ed. Taxonomy Binomial System Species Identification Classification Categories Phylogenetic Trees Tracing Phylogeny Cladistic Systematics
Outline Classification of Living Things Chapter 20 Mader: Biology 8th Ed. Taxonomy Binomial System Species Identification Classification Categories Phylogenetic Trees Tracing Phylogeny Cladistic Systematics
7. Tests for selection
 Sequence analysis and genomics 7. Tests for selection Dr. Katja Nowick Group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute for Brain Research www. nowicklab.info
Sequence analysis and genomics 7. Tests for selection Dr. Katja Nowick Group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute for Brain Research www. nowicklab.info
Integrative Biology 200 "PRINCIPLES OF PHYLOGENETICS" Spring 2018 University of California, Berkeley
 Integrative Biology 200 "PRINCIPLES OF PHYLOGENETICS" Spring 2018 University of California, Berkeley B.D. Mishler Feb. 14, 2018. Phylogenetic trees VI: Dating in the 21st century: clocks, & calibrations;
Integrative Biology 200 "PRINCIPLES OF PHYLOGENETICS" Spring 2018 University of California, Berkeley B.D. Mishler Feb. 14, 2018. Phylogenetic trees VI: Dating in the 21st century: clocks, & calibrations;
Evolutionary trees. Describe the relationship between objects, e.g. species or genes
 Evolutionary trees Bonobo Chimpanzee Human Neanderthal Gorilla Orangutan Describe the relationship between objects, e.g. species or genes Early evolutionary studies The evolutionary relationships between
Evolutionary trees Bonobo Chimpanzee Human Neanderthal Gorilla Orangutan Describe the relationship between objects, e.g. species or genes Early evolutionary studies The evolutionary relationships between
Evolu&on, Popula&on Gene&cs, and Natural Selec&on Computa.onal Genomics Seyoung Kim
 Evolu&on, Popula&on Gene&cs, and Natural Selec&on 02-710 Computa.onal Genomics Seyoung Kim Phylogeny of Mammals Phylogene&cs vs. Popula&on Gene&cs Phylogene.cs Assumes a single correct species phylogeny
Evolu&on, Popula&on Gene&cs, and Natural Selec&on 02-710 Computa.onal Genomics Seyoung Kim Phylogeny of Mammals Phylogene&cs vs. Popula&on Gene&cs Phylogene.cs Assumes a single correct species phylogeny
Phylogeny and systematics. Why are these disciplines important in evolutionary biology and how are they related to each other?
 Phylogeny and systematics Why are these disciplines important in evolutionary biology and how are they related to each other? Phylogeny and systematics Phylogeny: the evolutionary history of a species
Phylogeny and systematics Why are these disciplines important in evolutionary biology and how are they related to each other? Phylogeny and systematics Phylogeny: the evolutionary history of a species
"PRINCIPLES OF PHYLOGENETICS: ECOLOGY AND EVOLUTION" Integrative Biology 200B Spring 2009 University of California, Berkeley
 "PRINCIPLES OF PHYLOGENETICS: ECOLOGY AND EVOLUTION" Integrative Biology 200B Spring 2009 University of California, Berkeley B.D. Mishler Jan. 22, 2009. Trees I. Summary of previous lecture: Hennigian
"PRINCIPLES OF PHYLOGENETICS: ECOLOGY AND EVOLUTION" Integrative Biology 200B Spring 2009 University of California, Berkeley B.D. Mishler Jan. 22, 2009. Trees I. Summary of previous lecture: Hennigian
The Importance of Systema2cs and Rassenkreis. Reading: Willi Hennig
 The Importance of Systema2cs and Rassenkreis Reading: Throughout the class so far we seen that the distribu2on of an organism is the result of its biological history as well as geologic and clima4c history
The Importance of Systema2cs and Rassenkreis Reading: Throughout the class so far we seen that the distribu2on of an organism is the result of its biological history as well as geologic and clima4c history
Molecular Evolution & Phylogenetics Traits, phylogenies, evolutionary models and divergence time between sequences
 Molecular Evolution & Phylogenetics Traits, phylogenies, evolutionary models and divergence time between sequences Basic Bioinformatics Workshop, ILRI Addis Ababa, 12 December 2017 1 Learning Objectives
Molecular Evolution & Phylogenetics Traits, phylogenies, evolutionary models and divergence time between sequences Basic Bioinformatics Workshop, ILRI Addis Ababa, 12 December 2017 1 Learning Objectives
Michael Yaffe Lecture #5 (((A,B)C)D) Database Searching & Molecular Phylogenetics A B C D B C D
C)D) Database Searching & Molecular Phylogenetics A B C D B C D") 7.91 Lecture #5 Database Searching & Molecular Phylogenetics Michael Yaffe B C D B C D (((,B)C)D) Outline Distance Matrix Methods Neighbor-Joining Method and Related Neighbor Methods Maximum Likelihood
7.91 Lecture #5 Database Searching & Molecular Phylogenetics Michael Yaffe B C D B C D (((,B)C)D) Outline Distance Matrix Methods Neighbor-Joining Method and Related Neighbor Methods Maximum Likelihood
CHAPTERS 24-25: Evidence for Evolution and Phylogeny
 CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
Thanks to Paul Lewis and Joe Felsenstein for the use of slides
 Thanks to Paul Lewis and Joe Felsenstein for the use of slides Review Hennigian logic reconstructs the tree if we know polarity of characters and there is no homoplasy UPGMA infers a tree from a distance
Thanks to Paul Lewis and Joe Felsenstein for the use of slides Review Hennigian logic reconstructs the tree if we know polarity of characters and there is no homoplasy UPGMA infers a tree from a distance
Chapter 26: Phylogeny and the Tree of Life
 Chapter 26: Phylogeny and the Tree of Life 1. Key Concepts Pertaining to Phylogeny 2. Determining Phylogenies 3. Evolutionary History Revealed in Genomes 1. Key Concepts Pertaining to Phylogeny PHYLOGENY
Chapter 26: Phylogeny and the Tree of Life 1. Key Concepts Pertaining to Phylogeny 2. Determining Phylogenies 3. Evolutionary History Revealed in Genomes 1. Key Concepts Pertaining to Phylogeny PHYLOGENY
Biologists have used many approaches to estimating the evolutionary history of organisms and using that history to construct classifications.
 Phylogenetic Inference Biologists have used many approaches to estimating the evolutionary history of organisms and using that history to construct classifications. Willi Hennig developed d the techniques
Phylogenetic Inference Biologists have used many approaches to estimating the evolutionary history of organisms and using that history to construct classifications. Willi Hennig developed d the techniques
Preliminaries. Download PAUP* from: Tuesday, July 19, 16
 Preliminaries Download PAUP* from: http://people.sc.fsu.edu/~dswofford/paup_test 1 A model of the Boston T System 1 Idea from Paul Lewis A simpler model? 2 Why do models matter? Model-based methods including
Preliminaries Download PAUP* from: http://people.sc.fsu.edu/~dswofford/paup_test 1 A model of the Boston T System 1 Idea from Paul Lewis A simpler model? 2 Why do models matter? Model-based methods including
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Evolutionary Tree Analysis. Overview
 CSI/BINF 5330 Evolutionary Tree Analysis Young-Rae Cho Associate Professor Department of Computer Science Baylor University Overview Backgrounds Distance-Based Evolutionary Tree Reconstruction Character-Based
CSI/BINF 5330 Evolutionary Tree Analysis Young-Rae Cho Associate Professor Department of Computer Science Baylor University Overview Backgrounds Distance-Based Evolutionary Tree Reconstruction Character-Based
Principles of Phylogeny Reconstruction How do we reconstruct the tree of life? Basic Terminology. Looking at Trees. Basic Terminology.
 Principles of Phylogeny Reconstruction How do we reconstruct the tree of life? Phylogeny: asic erminology Outline: erminology Phylogenetic tree: Methods Problems parsimony maximum likelihood bootstrapping
Principles of Phylogeny Reconstruction How do we reconstruct the tree of life? Phylogeny: asic erminology Outline: erminology Phylogenetic tree: Methods Problems parsimony maximum likelihood bootstrapping