Taxonomical Classification using:
|
|
|
- Marcus Arnold
- 5 years ago
- Views:
Transcription

1 Taxonomical Classification using: Extracting ecological signal from noise: introduction to tools for the analysis of NGS data from microbial communities Bergen, April
2 INTRODUCTION Taxonomical prediction = Who is out there and how many? Composition of the microbial community SSU rrna (16S/18S) - de facto standard in environmental genomics Amplicons or rrna tags Shotgun rrna / RNA-Seq (LSU + SSU) Classification of subset (~0.1%) in shotgun metagenome data
3 INTRODUCTION Taxonomy = system for classification Phylogeny = evolutionary development Bad phylogeny -> bad taxonomy Bad taxonomy -> Bad / less meaningful classification
4 AVAILABLE TAXONOMIES AND REF. DATABASES NCBI Taxonomy. Not meant to be authoritative but what sequences in Genbank are mapped to. Commonly used for taxonomical classification (best hit, MEGAN) Polyphyletic unclassified nodes, or even incorrectly. Incorrect assignments and expired taxa. RDP (Ribosomal Database Project) Greengenes --> SILVA <--
5 SILVA Includes all three domains of life (including Eukaryotes) SSURef 106: ~500k full length SSU sequences and 20k LSU sequences Taxonomy assignments to clusters that include uncultured organisms (up to genus level) Distributed for the ARB software packages, plus some online resources
6 CLASSIFICATION METHODS Can be roughly divided into those based on: 1. Inferred multiple alignments (e.g. NAST) 2. Nucleotide composition (e.g. RDP Classifier) 3. Pairwise alignments (e.g. BLAST)
7 CLASSIFICATION METHODS 1. Infer multiple alignment (NAST, SINA WebAligner, etc) and insert into existing reference tree (GreenGenes classifier, LCA) + Best accuracy for reads close to known reference sequences [Liu et al, 2008] - Slow and sensitive to read novelty or quality
8 CLASSIFICATION METHODS 2. Nucleotide composition based - RDP Classifier (8-mer): + Fast. Similar results to BLAST in environmental datasets [Liu et al, 2008] - More sensitive to sequencing noise and small differences 3. Pairwise alignment to reference database (Best BLAST hit, Lowest Common Ancestor, MEGAN) + With LCA relatively fast and accurate [Liu et al, 2008] - LCA very sensitive to assignments in ref. database
9 CLASSIFICATION METHODS In addition: Methods based on reconstruction of phylogenetic tree. + Ability to study phylogenetic novelty - Slow and expensive - High false positive-rate in Liu et al benchmark
10
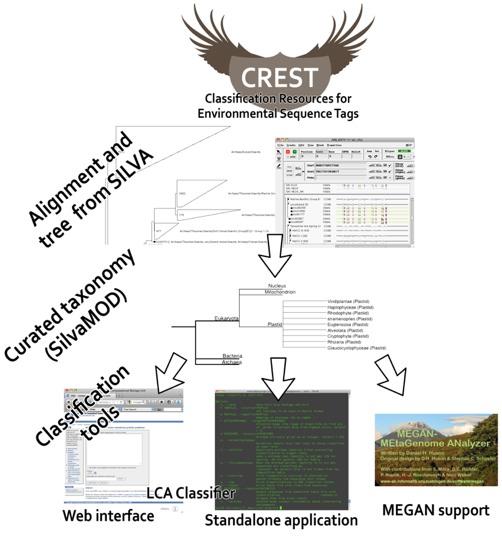
11 CREST WORKFLOW Alignment (Megablast) to the SilvaMod reference database and LCA using custom python script or MEGAN [Huson et al, 2007]. Mapping taxa to ranks using NCBI Taxonomy Minimum similarity filters (99% for species, 97% for genus, 95% for family, 90% for order...) Web interface (max. 1,000 sequences) including Megablast (under development using Hodman)
12 2% range from top Scoring BLAST Hit, min score=155 bits Blast match #1, Score = 100 bits Query: 1 CTGCCCTGGCTTCTATTATGCGTGACGT... Sbjct: 350 CTGCCCGGGC-TCTATTATGCGTGACGT... Blast match #2, Score = 95 bits Query: 1 CTGCCCTGGCTTCTATTATGCGTGACGT... Sbjct: 349 CTGCCCGGGC--CTATTAGGCGTGACGT... Blast match #3, Score = 90 bits Query: 3 CCCTGGCTTCTATTA-TGCGTGACGTGTC... Sbjct: 353 CCCGTGC-TCTATTAGTGCGTGACCTATG...
13 OUTPUT /*0'$()(*1* 2*1*!34$+*$5' 67*0' 8$#94' :7*, :'--4-*0),0%*$!057*'* =><<=C? D<E ;<C :'--4-*0),0%*$F4G*0H,(* A =>===AA A I,$' 2,(*-?<=A =><<<CC JA; 8$5-*""#K#'+)*()+,&*#$)-'.'- D =>===DJ A!""#$%&'$(")*()+,&*#$)-'.'-!""#$%&'$(")*()L7H-4&)-'.'- /*0'$()(*1* 2*1*!34$+*$5' 67*0' 8$#94' :7*,!057*'* 27*4&*057*',(* J I,$'!057*'* F40H*057*',(* ;E =>==?J@ B*5('0#*!5#+,3*5('0#*? I,$' B*5('0#*!5(#$,3*5('0#* J I,$' B*5('0#* B*5('0,#+'('" JEC =>=EA@A ;A A@E B*5('0#* BMANE E =>===EC J? B*5('0#* :*-+#(70#1 AE =>==AC? A I,$' B*5('0#* :7-,0,3# E =>===EC J? B*5('0#* :H*$,3*5('0#* A =>===AA A I,$' Also FASTA format with assignments for each sequence + a more parser-friendly format for abundance.
14 PERFORMANCE TESTING Exhaustive tenfold cross validation: aligning 1/10 of reference database to the other 9/10 Different lengths (full-length, 450 bp and 100 bp) Gives recall rate and false positive rate Removal of taxa: cross validation removing whole genera, families or phyla and aligning to remaining Real data: Assignment of 4 different SSU rrna datasets from environmental genomics studies
15 COMPARISON TO OTHER METHODS Greengenes Similar approach used very recently to create alignment-informed consensus taxonomy Larger database, but few sequences annotated to genus rank Alternative files for LCA classification built RDP Classifier Nucleotide composition based + Naïve Bayes Classifier Used with default training set + Greengenes (QIIME)
16 RESULTS ROC for 10 split cross validation, Family rank (Fragment length=450bp) ROC for 10 split cross validation, Genus rank (Fragment length=450bp) Recall rate SilvaMod106/LCA Greengenes/LCA Greengenes/RDP Classifier RDP Classifier default LCA range=0.02 Confidence cutoff=0.8 Recall rate SilvaMod106/LCA Greengenes/LCA Greengenes/RDP Classifier RDP Classifier default LCA range=0.02 Confidence cutoff= False Positive Rate False Positive Rate
17 RESULTS ROC for 10 split cross validation, Family rank (Fragment length=100bp) ROC for 10 split cross validation, Genus rank (Fragment length=100bp) Recall rate SilvaMod106/LCA Greengenes/LCA Greengenes/RDP Classifier RDP Classifier default LCA range=0.02 Confidence cutoff=0.8 Recall rate SilvaMod106/LCA Greengenes/LCA Greengenes/RDP Classifier RDP Classifier default LCA range=0.02 Confidence cutoff= False Positive Rate False Positive Rate
18 RESULTS!"#$%&'(./"0%)).%#1.2%)*".34*(5(6".$%5".2$4'.$"'46%)74275%8%. 0$4**.6%)(1%5(4#9. :"5,41 &%)*".+4*(5(6"./%5".%5. ;$%(#(#<.=. &$%<'"#5. $"'46"1.$%#>.)"6").24$. /"2"$"#0".*"5 )"#<5,!"#"$% &%'()("* +,-)%!"# $ %&'($)*+,-!-. /-/01 /-/22 /-23!"# $ %&'($)*+ 45/678 /-93 /-31 /-:3!"# $ %&'($)*+ 0//678 /-02 /-34 /-94!"# $ ;<==>?=>=@,-!-. /-05 /-//0: /-13!"# $ ;<==>?=>=@ 45/678 /-0/ /-05 /-:1!"# $ ;<==>?=>=@ 0//678 /-03 /-9A /-92 BCD 7 ;<==>?=>=@,-!-. E /-43 /-05 BCD 7 ;<==>?=>=@ 45/678 E /-0: /-/A3 BCD 7 ;<==>?=>=@ 0//678 E /-//:9 /-/04 BCD 7 BCD6(1,-!-. /-3A /-3A /-:2 BCD 7 BCD6(1 45/678 /-95 /-99 /-00 BCD 7 BCD6(1 0//678 /-/1A /-/:: /-/3: False Positive Rate Removal of whole families cross validation (Fragment length=450bp, SilvaMod106+LCA) Genus rank Family rank Phylum rank $!"#6.'$@@&G&.$I&*>R66H@&>?6)=?$7'$@I6$'&?>L=>I@6Q&IJ&>6$69S6<$>?=6*G6 IJ=6J&?J=@I67&I@.*<=66$@6Q=''6$@68=<.=>I6@&L&'$<&IP6G&'I=<@ Relative LCA range 7 M$N(=6O$P=@6.'$@@&G&.$I&*>6H@&>?6IJ=6BCD6"'$@@&G&=<6Q&IJ6$67**I@I<$86.*>G&+=>.=6.HI*GG6*G6/-A. F>E.<*88=+6GH''E'=>?IJ6@=KH=>.=@6G<*L6IJ=6<=G=<=>.=6*<6I<$&>&>?6+$I$@=
19 RESULTS A2*20"* %.67#%.-(78 $4879:#94(* "(178(.-#94(* AB584:C78,.*#,.:!"#$"%&'%() *"&+%,-,(.!"#$%&#'(!"#$%&#'( )**+,(-.!"#$%&#'( U L7.59#T(:C#.#V%0"'P#.*(E-,7-:#1(:9O487#W#FJ#:4#.#97X+7-O7#(-#"(*2.345 E'F525.)*.G" "C4:E+-#,7:.E7-4,7 "C4:E+-#,7:.:8.-9O8(Q:4,7 =G"#8LP0#.,Q*(O4-9 =G"#8LP0#.,Q*(O4-9 B,*2-)!!9)5DH>) 5"270 I FFI F=S?J??S=K< ISDJ< 6"*+,7 B52'%'%()C)!+25"),8)5"270)200'(%"7 A2*20"* 9%'#$")*2:2);<=>=?@ 2 D"8"5"%&")0"* /"%$0 123' $3 /"%"52 123'-'" %/0 1 "(*2.345 %.67#%.-(78!"#$% &!#&% ''#(%!)*+*),(*+*$ ))*+*$ %/0 1 "(*2.345 $4879:#94(*!+#,% "'#-% ''#)% $!$*+*)"" )(";);)"& $'*$*," %/0 1 "(*2.345 "(178(.-#94(* <=>?@ "'#(% '!#"% ()*)*+ -)*)*+ $+*)*+ %/0 1 "(*2.345 AB584:C78,.*#,.: -&#(% '!#+% ''#"%!"*$*),$*-*) =D;?;= %/0 1!877-E7-79 %.67#%.-(78 ==>F@ GH>F@ DI>D@ =F;J;J?F;J;J )!*+*+ %/0 1!877-E7-79 $4879:#94(* =H>K@ FF>=@ IH>=@ =<J;J;J =?G;=;J <=;?;? %/0 1!877-E7-79 "(178(.-#94(*!'#+% GJ>=@ IF>G@ <I;=;J F<;=;J =I;=;J %/0 1!877-E7-79 AB584:C78,.*#,.: KK>F@ ID>J@ DD>H@ =F;=;J?<;G;J $)*$*) LMN O!877-E7-79 %.67#%.-(78 J K?>?@ D=>I@ J?I;J;J D;J;J LMN O!877-E7-79 $4879:#94(* J F?>?@ IG>K@ J ===;J;J =G;?;= LMN O!877-E7-79 "(178(.-#94(* J F<>H@ DJ>F@ J F<;=;J =J;=;J LMN O!877-E7-79 AB584:C78,.*#,.: J I=>G@ DK>I@ J =D;<;J D;?;J LMN O LMN#2G %.67#%.-(78 D><@ F=>=@ IK>=@ =K;J;J?J;J;? =J;J;? LMN O LMN#2G $4879:#94(* ==>D@ HJ>H@ IJ>D@ =KG;?;J DF;?;J?J;?;= LMN O LMN#2G "(178(.-#94(* G>K@ <D>K@ GG>J@ <G;=;J <D;=;J =J;=;J LMN O LMN#2G AB584:C78,.*#,.: IH>H@ D=>K@ DK>K@?=;?;J =K;?;J I;?;J. P+,178#4R#+-(X+7#:.Y.#E(27-#97Q.8.:7*B#R48#1.O:78(.#;#.8OC.7.#;#7+6.8B4:79>#ZC787#:C7#C(EC79:#:4:.*#-+,178#4R#:.Y.#
20 RESULTS


21 ACKNOWLEDGMENTS Tim Urich Steffen Jørgensen Lise Øvreås Inge Jonassen Daniel Huson Markus Gorfer Svenn Helge Grindhaug
Robert Edgar. Independent scientist
 Robert Edgar Independent scientist robert@drive5.com www.drive5.com "Bacterial taxonomy is a hornets nest that no one, really, wants to get into." Referee #1, UTAX paper Assume prokaryotic species meaningful
Robert Edgar Independent scientist robert@drive5.com www.drive5.com "Bacterial taxonomy is a hornets nest that no one, really, wants to get into." Referee #1, UTAX paper Assume prokaryotic species meaningful
Assigning Taxonomy to Marker Genes. Susan Huse Brown University August 7, 2014
 Assigning Taxonomy to Marker Genes Susan Huse Brown University August 7, 2014 In a nutshell Taxonomy is assigned by comparing your DNA sequences against a database of DNA sequences from known taxa Marker
Assigning Taxonomy to Marker Genes Susan Huse Brown University August 7, 2014 In a nutshell Taxonomy is assigned by comparing your DNA sequences against a database of DNA sequences from known taxa Marker
Comparison of Three Fugal ITS Reference Sets. Qiong Wang and Jim R. Cole
 RDP TECHNICAL REPORT Created 04/12/2014, Updated 08/08/2014 Summary Comparison of Three Fugal ITS Reference Sets Qiong Wang and Jim R. Cole wangqion@msu.edu, colej@msu.edu In this report, we evaluate the
RDP TECHNICAL REPORT Created 04/12/2014, Updated 08/08/2014 Summary Comparison of Three Fugal ITS Reference Sets Qiong Wang and Jim R. Cole wangqion@msu.edu, colej@msu.edu In this report, we evaluate the
Microbiome: 16S rrna Sequencing 3/30/2018
 Microbiome: 16S rrna Sequencing 3/30/2018 Skills from Previous Lectures Central Dogma of Biology Lecture 3: Genetics and Genomics Lecture 4: Microarrays Lecture 12: ChIP-Seq Phylogenetics Lecture 13: Phylogenetics
Microbiome: 16S rrna Sequencing 3/30/2018 Skills from Previous Lectures Central Dogma of Biology Lecture 3: Genetics and Genomics Lecture 4: Microarrays Lecture 12: ChIP-Seq Phylogenetics Lecture 13: Phylogenetics
A Bayesian taxonomic classification method for 16S rrna gene sequences with improved species-level accuracy
 Gao et al. BMC Bioinformatics (2017) 18:247 DOI 10.1186/s12859-017-1670-4 SOFTWARE Open Access A Bayesian taxonomic classification method for 16S rrna gene sequences with improved species-level accuracy
Gao et al. BMC Bioinformatics (2017) 18:247 DOI 10.1186/s12859-017-1670-4 SOFTWARE Open Access A Bayesian taxonomic classification method for 16S rrna gene sequences with improved species-level accuracy
Taxonomy and Clustering of SSU rrna Tags. Susan Huse Josephine Bay Paul Center August 5, 2013
 Taxonomy and Clustering of SSU rrna Tags Susan Huse Josephine Bay Paul Center August 5, 2013 Primary Methods of Taxonomic Assignment Bayesian Kmer Matching RDP http://rdp.cme.msu.edu Wang, et al (2007)
Taxonomy and Clustering of SSU rrna Tags Susan Huse Josephine Bay Paul Center August 5, 2013 Primary Methods of Taxonomic Assignment Bayesian Kmer Matching RDP http://rdp.cme.msu.edu Wang, et al (2007)
Title ghost-tree: creating hybrid-gene phylogenetic trees for diversity analyses
 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 Title ghost-tree: creating hybrid-gene phylogenetic trees for diversity analyses
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 Title ghost-tree: creating hybrid-gene phylogenetic trees for diversity analyses
08/21/2017 BLAST. Multiple Sequence Alignments: Clustal Omega
 BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
MiGA: The Microbial Genome Atlas
 December 12 th 2017 MiGA: The Microbial Genome Atlas Jim Cole Center for Microbial Ecology Dept. of Plant, Soil & Microbial Sciences Michigan State University East Lansing, Michigan U.S.A. Where I m From
December 12 th 2017 MiGA: The Microbial Genome Atlas Jim Cole Center for Microbial Ecology Dept. of Plant, Soil & Microbial Sciences Michigan State University East Lansing, Michigan U.S.A. Where I m From
An Automated Phylogenetic Tree-Based Small Subunit rrna Taxonomy and Alignment Pipeline (STAP)
") An Automated Phylogenetic Tree-Based Small Subunit rrna Taxonomy and Alignment Pipeline (STAP) Dongying Wu 1 *, Amber Hartman 1,6, Naomi Ward 4,5, Jonathan A. Eisen 1,2,3 1 UC Davis Genome Center, University
An Automated Phylogenetic Tree-Based Small Subunit rrna Taxonomy and Alignment Pipeline (STAP) Dongying Wu 1 *, Amber Hartman 1,6, Naomi Ward 4,5, Jonathan A. Eisen 1,2,3 1 UC Davis Genome Center, University
Nature Biotechnology: doi: /nbt Supplementary Figure 1. Detailed overview of the primer-free full-length SSU rrna library preparation.
 Supplementary Figure 1 Detailed overview of the primer-free full-length SSU rrna library preparation. Detailed overview of the primer-free full-length SSU rrna library preparation. Supplementary Figure
Supplementary Figure 1 Detailed overview of the primer-free full-length SSU rrna library preparation. Detailed overview of the primer-free full-length SSU rrna library preparation. Supplementary Figure
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Prac%cal Bioinforma%cs for Life Scien%sts. Week 14, Lecture 28. István Albert Bioinforma%cs Consul%ng Center Penn State
 Prac%cal Bioinforma%cs for Life Scien%sts Week 14, Lecture 28 István Albert Bioinforma%cs Consul%ng Center Penn State Final project A group of researchers are interested in studying protein binding loca%ons
Prac%cal Bioinforma%cs for Life Scien%sts Week 14, Lecture 28 István Albert Bioinforma%cs Consul%ng Center Penn State Final project A group of researchers are interested in studying protein binding loca%ons
Accuracy of taxonomy prediction for 16S rrna and fungal ITS sequences
 Accuracy of taxonomy prediction for 16S rrna and fungal ITS sequences Robert C. Edgar Sonoma, CA, USA ABSTRACT Prediction of taxonomy for marker gene sequences such as 16S ribosomal RNA (rrna) is a fundamental
Accuracy of taxonomy prediction for 16S rrna and fungal ITS sequences Robert C. Edgar Sonoma, CA, USA ABSTRACT Prediction of taxonomy for marker gene sequences such as 16S ribosomal RNA (rrna) is a fundamental
SUPPLEMENTARY INFORMATION
 Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Heuristic Alignment and Searching
 3/28/2012 Types of alignments Global Alignment Each letter of each sequence is aligned to a letter or a gap (e.g., Needleman-Wunsch). Local Alignment An optimal pair of subsequences is taken from the two
3/28/2012 Types of alignments Global Alignment Each letter of each sequence is aligned to a letter or a gap (e.g., Needleman-Wunsch). Local Alignment An optimal pair of subsequences is taken from the two
SUPPLEMENTARY INFORMATION
 Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
BLAST. Varieties of BLAST
 BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
Centrifuge: rapid and sensitive classification of metagenomic sequences
 Centrifuge: rapid and sensitive classification of metagenomic sequences Daehwan Kim, Li Song, Florian P. Breitwieser, and Steven L. Salzberg Supplementary Material Supplementary Table 1 Supplementary Note
Centrifuge: rapid and sensitive classification of metagenomic sequences Daehwan Kim, Li Song, Florian P. Breitwieser, and Steven L. Salzberg Supplementary Material Supplementary Table 1 Supplementary Note
Amplicon Sequencing. Dr. Orla O Sullivan SIRG Research Fellow Teagasc
 Amplicon Sequencing Dr. Orla O Sullivan SIRG Research Fellow Teagasc What is Amplicon Sequencing? Sequencing of target genes (are regions of ) obtained by PCR using gene specific primers. Why do we do
Amplicon Sequencing Dr. Orla O Sullivan SIRG Research Fellow Teagasc What is Amplicon Sequencing? Sequencing of target genes (are regions of ) obtained by PCR using gene specific primers. Why do we do
The Effect of Primer Choice and Short Read Sequences on the Outcome of 16S rrna Gene Based Diversity Studies
 The Effect of Primer Choice and Short Read Sequences on the Outcome of 16S rrna Gene Based Diversity Studies Jonas Ghyselinck 1 *., Stefan Pfeiffer 2 *., Kim Heylen 1, Angela Sessitsch 2, Paul De Vos 1
The Effect of Primer Choice and Short Read Sequences on the Outcome of 16S rrna Gene Based Diversity Studies Jonas Ghyselinck 1 *., Stefan Pfeiffer 2 *., Kim Heylen 1, Angela Sessitsch 2, Paul De Vos 1
Grundlagen der Bioinformatik Summer semester Lecturer: Prof. Daniel Huson
 Grundlagen der Bioinformatik, SS 10, D. Huson, April 12, 2010 1 1 Introduction Grundlagen der Bioinformatik Summer semester 2010 Lecturer: Prof. Daniel Huson Office hours: Thursdays 17-18h (Sand 14, C310a)
Grundlagen der Bioinformatik, SS 10, D. Huson, April 12, 2010 1 1 Introduction Grundlagen der Bioinformatik Summer semester 2010 Lecturer: Prof. Daniel Huson Office hours: Thursdays 17-18h (Sand 14, C310a)
A (short) introduction to phylogenetics
 introduction to phylogenetics") A (short) introduction to phylogenetics Thibaut Jombart, Marie-Pauline Beugin MRC Centre for Outbreak Analysis and Modelling Imperial College London Genetic data analysis with PR Statistics, Millport Field
A (short) introduction to phylogenetics Thibaut Jombart, Marie-Pauline Beugin MRC Centre for Outbreak Analysis and Modelling Imperial College London Genetic data analysis with PR Statistics, Millport Field
PHYLOGENY AND SYSTEMATICS
 AP BIOLOGY EVOLUTION/HEREDITY UNIT Unit 1 Part 11 Chapter 26 Activity #15 NAME DATE PERIOD PHYLOGENY AND SYSTEMATICS PHYLOGENY Evolutionary history of species or group of related species SYSTEMATICS Study
AP BIOLOGY EVOLUTION/HEREDITY UNIT Unit 1 Part 11 Chapter 26 Activity #15 NAME DATE PERIOD PHYLOGENY AND SYSTEMATICS PHYLOGENY Evolutionary history of species or group of related species SYSTEMATICS Study
Microbes usually have few distinguishing properties that relate them, so a hierarchical taxonomy mainly has not been possible.
 Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Microbial Taxonomy. Slowly evolving molecules (e.g., rrna) used for large-scale structure; "fast- clock" molecules for fine-structure.
 used for large-scale structure; fast- clock molecules for fine-structure.") Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Handling Fungal data in MoBeDAC
 Handling Fungal data in MoBeDAC Jason Stajich UC Riverside Fungal Taxonomy and naming undergoing a revolution One fungus, one name http://www.biology.duke.edu/fungi/ mycolab/primers.htm http://www.biology.duke.edu/fungi/
Handling Fungal data in MoBeDAC Jason Stajich UC Riverside Fungal Taxonomy and naming undergoing a revolution One fungus, one name http://www.biology.duke.edu/fungi/ mycolab/primers.htm http://www.biology.duke.edu/fungi/
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
BLAST Database Searching. BME 110: CompBio Tools Todd Lowe April 8, 2010
 BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
objective functions...
 objective functions... COFFEE (Notredame et al. 1998) measures column by column similarity between pairwise and multiple sequence alignments assumes that the pairwise alignments are optimal assumes a set
objective functions... COFFEE (Notredame et al. 1998) measures column by column similarity between pairwise and multiple sequence alignments assumes that the pairwise alignments are optimal assumes a set
Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics
 Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics Tandy Warnow Founder Professor of Engineering The University of Illinois at Urbana-Champaign http://tandy.cs.illinois.edu
Using Ensembles of Hidden Markov Models for Grand Challenges in Bioinformatics Tandy Warnow Founder Professor of Engineering The University of Illinois at Urbana-Champaign http://tandy.cs.illinois.edu
Supplemental Online Results:
 Supplemental Online Results: Functional, phylogenetic, and computational determinants of prediction accuracy using reference genomes A series of tests determined the relationship between PICRUSt s prediction
Supplemental Online Results: Functional, phylogenetic, and computational determinants of prediction accuracy using reference genomes A series of tests determined the relationship between PICRUSt s prediction
PGA: A Program for Genome Annotation by Comparative Analysis of. Maximum Likelihood Phylogenies of Genes and Species
 PGA: A Program for Genome Annotation by Comparative Analysis of Maximum Likelihood Phylogenies of Genes and Species Paulo Bandiera-Paiva 1 and Marcelo R.S. Briones 2 1 Departmento de Informática em Saúde
PGA: A Program for Genome Annotation by Comparative Analysis of Maximum Likelihood Phylogenies of Genes and Species Paulo Bandiera-Paiva 1 and Marcelo R.S. Briones 2 1 Departmento de Informática em Saúde
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Microbial Taxonomy. Microbes usually have few distinguishing properties that relate them, so a hierarchical taxonomy mainly has not been possible.
 Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Other resources. Greengenes (bacterial) Silva (bacteria, archaeal and eukarya)
 Silva (bacteria, archaeal and eukarya)") General QIIME resources http://qiime.org/ Blog (news, updates): http://qiime.wordpress.com/ Support/forum: https://groups.google.com/forum/#!forum/qiimeforum Citing QIIME: Caporaso, J.G. et al., QIIME
General QIIME resources http://qiime.org/ Blog (news, updates): http://qiime.wordpress.com/ Support/forum: https://groups.google.com/forum/#!forum/qiimeforum Citing QIIME: Caporaso, J.G. et al., QIIME
rrdp: Interface to the RDP Classifier
 rrdp: Interface to the RDP Classifier Michael Hahsler Anurag Nagar Abstract This package installs and interfaces the naive Bayesian classifier for 16S rrna sequences developed by the Ribosomal Database
rrdp: Interface to the RDP Classifier Michael Hahsler Anurag Nagar Abstract This package installs and interfaces the naive Bayesian classifier for 16S rrna sequences developed by the Ribosomal Database
Comparing whole genomes
 BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
Introduction to microbiota data analysis
 Introduction to microbiota data analysis Natalie Knox, PhD Head Bacterial Genomics, Bioinformatics Core National Microbiology Laboratory, Public Health Agency of Canada 2 National Microbiology Laboratory
Introduction to microbiota data analysis Natalie Knox, PhD Head Bacterial Genomics, Bioinformatics Core National Microbiology Laboratory, Public Health Agency of Canada 2 National Microbiology Laboratory
2MHR. Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity.
 Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Microbial Taxonomy and the Evolution of Diversity
 19 Microbial Taxonomy and the Evolution of Diversity Copyright McGraw-Hill Global Education Holdings, LLC. Permission required for reproduction or display. 1 Taxonomy Introduction to Microbial Taxonomy
19 Microbial Taxonomy and the Evolution of Diversity Copyright McGraw-Hill Global Education Holdings, LLC. Permission required for reproduction or display. 1 Taxonomy Introduction to Microbial Taxonomy
Microbes and you ON THE LATEST HUMAN MICROBIOME DISCOVERIES, COMPUTATIONAL QUESTIONS AND SOME SOLUTIONS. Elizabeth Tseng
 Microbes and you ON THE LATEST HUMAN MICROBIOME DISCOVERIES, COMPUTATIONAL QUESTIONS AND SOME SOLUTIONS Elizabeth Tseng Dept. of CSE, University of Washington Johanna Lampe Lab, Fred Hutchinson Cancer
Microbes and you ON THE LATEST HUMAN MICROBIOME DISCOVERIES, COMPUTATIONAL QUESTIONS AND SOME SOLUTIONS Elizabeth Tseng Dept. of CSE, University of Washington Johanna Lampe Lab, Fred Hutchinson Cancer
1 Abstract. 2 Introduction. 3 Requirements. 4 Procedure
 1 Abstract None 2 Introduction The archaeal core set is used in testing the completeness of the archaeal draft genomes. The core set comprises of conserved single copy genes from 25 genomes. Coverage statistic
1 Abstract None 2 Introduction The archaeal core set is used in testing the completeness of the archaeal draft genomes. The core set comprises of conserved single copy genes from 25 genomes. Coverage statistic
Single alignment: Substitution Matrix. 16 march 2017
 Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction. Lesser Tenrec (Echinops telfairi)
") Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction Lesser Tenrec (Echinops telfairi) Goals: 1. Use phylogenetic experimental design theory to select optimal taxa to
Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction Lesser Tenrec (Echinops telfairi) Goals: 1. Use phylogenetic experimental design theory to select optimal taxa to
Bacterial Communities in Women with Bacterial Vaginosis: High Resolution Phylogenetic Analyses Reveal Relationships of Microbiota to Clinical Criteria
 Bacterial Communities in Women with Bacterial Vaginosis: High Resolution Phylogenetic Analyses Reveal Relationships of Microbiota to Clinical Criteria Seminar presentation Pierre Barbera Supervised by:
Bacterial Communities in Women with Bacterial Vaginosis: High Resolution Phylogenetic Analyses Reveal Relationships of Microbiota to Clinical Criteria Seminar presentation Pierre Barbera Supervised by:
Taxonomy. Content. How to determine & classify a species. Phylogeny and evolution
 Taxonomy Content Why Taxonomy? How to determine & classify a species Domains versus Kingdoms Phylogeny and evolution Why Taxonomy? Classification Arrangement in groups or taxa (taxon = group) Nomenclature
Taxonomy Content Why Taxonomy? How to determine & classify a species Domains versus Kingdoms Phylogeny and evolution Why Taxonomy? Classification Arrangement in groups or taxa (taxon = group) Nomenclature
Introduction to Evolutionary Concepts
 Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2009 VMD/MultiSeq
Introduction to Evolutionary Concepts and VMD/MultiSeq - Part I Zaida (Zan) Luthey-Schulten Dept. Chemistry, Beckman Institute, Biophysics, Institute of Genomics Biology, & Physics NIH Workshop 2009 VMD/MultiSeq
Microbial analysis with STAMP
 Microbial analysis with STAMP Conor Meehan cmeehan@itg.be A quick aside on who I am Tangents already! Who I am A postdoc at the Institute of Tropical Medicine in Antwerp, Belgium Mycobacteria evolution
Microbial analysis with STAMP Conor Meehan cmeehan@itg.be A quick aside on who I am Tangents already! Who I am A postdoc at the Institute of Tropical Medicine in Antwerp, Belgium Mycobacteria evolution
Bioinformatics tools for phylogeny and visualization. Yanbin Yin
 Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
Phylogenetic analyses. Kirsi Kostamo
 Phylogenetic analyses Kirsi Kostamo The aim: To construct a visual representation (a tree) to describe the assumed evolution occurring between and among different groups (individuals, populations, species,
Phylogenetic analyses Kirsi Kostamo The aim: To construct a visual representation (a tree) to describe the assumed evolution occurring between and among different groups (individuals, populations, species,
Using Bioinformatics to Study Evolutionary Relationships Instructions
 3 Using Bioinformatics to Study Evolutionary Relationships Instructions Student Researcher Background: Making and Using Multiple Sequence Alignments One of the primary tasks of genetic researchers is comparing
3 Using Bioinformatics to Study Evolutionary Relationships Instructions Student Researcher Background: Making and Using Multiple Sequence Alignments One of the primary tasks of genetic researchers is comparing
Genomics and bioinformatics summary. Finding genes -- computer searches
 Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Genomics and bioinformatics summary 1. Gene finding: computer searches, cdnas, ESTs, 2. Microarrays 3. Use BLAST to find homologous sequences 4. Multiple sequence alignments (MSAs) 5. Trees quantify sequence
Grundlagen der Bioinformatik, SS 08, D. Huson, May 2,
 Grundlagen der Bioinformatik, SS 08, D. Huson, May 2, 2008 39 5 Blast This lecture is based on the following, which are all recommended reading: R. Merkl, S. Waack: Bioinformatik Interaktiv. Chapter 11.4-11.7
Grundlagen der Bioinformatik, SS 08, D. Huson, May 2, 2008 39 5 Blast This lecture is based on the following, which are all recommended reading: R. Merkl, S. Waack: Bioinformatik Interaktiv. Chapter 11.4-11.7
Sequencing alignment Ameer Effat M. Elfarash
 Sequencing alignment Ameer Effat M. Elfarash Dept. of Genetics Fac. of Agriculture, Assiut Univ. aelfarash@aun.edu.eg Why perform a multiple sequence alignment? MSAs are at the heart of comparative genomics
Sequencing alignment Ameer Effat M. Elfarash Dept. of Genetics Fac. of Agriculture, Assiut Univ. aelfarash@aun.edu.eg Why perform a multiple sequence alignment? MSAs are at the heart of comparative genomics
Biological Networks: Comparison, Conservation, and Evolution via Relative Description Length By: Tamir Tuller & Benny Chor
 Biological Networks:,, and via Relative Description Length By: Tamir Tuller & Benny Chor Presented by: Noga Grebla Content of the presentation Presenting the goals of the research Reviewing basic terms
Biological Networks:,, and via Relative Description Length By: Tamir Tuller & Benny Chor Presented by: Noga Grebla Content of the presentation Presenting the goals of the research Reviewing basic terms
Synteny Portal Documentation
 Synteny Portal Documentation Synteny Portal is a web application portal for visualizing, browsing, searching and building synteny blocks. Synteny Portal provides four main web applications: SynCircos,
Synteny Portal Documentation Synteny Portal is a web application portal for visualizing, browsing, searching and building synteny blocks. Synteny Portal provides four main web applications: SynCircos,
In-Depth Assessment of Local Sequence Alignment
 2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
BINF6201/8201. Molecular phylogenetic methods
 BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
Overview - MS Proteomics in One Slide. MS masses of peptides. MS/MS fragments of a peptide. Results! Match to sequence database
 Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
Basic Local Alignment Search Tool
 Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
C3020 Molecular Evolution. Exercises #3: Phylogenetics
 C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
Chapter 26 Phylogeny and the Tree of Life
 Chapter 26 Phylogeny and the Tree of Life Chapter focus Shifting from the process of how evolution works to the pattern evolution produces over time. Phylogeny Phylon = tribe, geny = genesis or origin
Chapter 26 Phylogeny and the Tree of Life Chapter focus Shifting from the process of how evolution works to the pattern evolution produces over time. Phylogeny Phylon = tribe, geny = genesis or origin
Macroevolution Part I: Phylogenies
 Macroevolution Part I: Phylogenies Taxonomy Classification originated with Carolus Linnaeus in the 18 th century. Based on structural (outward and inward) similarities Hierarchal scheme, the largest most
Macroevolution Part I: Phylogenies Taxonomy Classification originated with Carolus Linnaeus in the 18 th century. Based on structural (outward and inward) similarities Hierarchal scheme, the largest most
RGP finder: prediction of Genomic Islands
 Training courses on MicroScope platform RGP finder: prediction of Genomic Islands Dynamics of bacterial genomes Gene gain Horizontal gene transfer Gene loss Deletion of one or several genes Duplication
Training courses on MicroScope platform RGP finder: prediction of Genomic Islands Dynamics of bacterial genomes Gene gain Horizontal gene transfer Gene loss Deletion of one or several genes Duplication
A multi-source domain annotation pipeline for quantitative metagenomic and metatranscriptomic functional profiling
 A multi-source domain annotation pipeline for quantitative metagenomic and metatranscriptomic functional profiling Ari Ugarte, Riccardo Vicedomini, Juliana Silva Bernardes, Alessandra Carbone 9 September,
A multi-source domain annotation pipeline for quantitative metagenomic and metatranscriptomic functional profiling Ari Ugarte, Riccardo Vicedomini, Juliana Silva Bernardes, Alessandra Carbone 9 September,
Constructing Evolutionary/Phylogenetic Trees
 Constructing Evolutionary/Phylogenetic Trees 2 broad categories: istance-based methods Ultrametric Additive: UPGMA Transformed istance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
Constructing Evolutionary/Phylogenetic Trees 2 broad categories: istance-based methods Ultrametric Additive: UPGMA Transformed istance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
Session 5: Phylogenomics
 Session 5: Phylogenomics B.- Phylogeny based orthology assignment REMINDER: Gene tree reconstruction is divided in three steps: homology search, multiple sequence alignment and model selection plus tree
Session 5: Phylogenomics B.- Phylogeny based orthology assignment REMINDER: Gene tree reconstruction is divided in three steps: homology search, multiple sequence alignment and model selection plus tree
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Overview of IslandPick pipeline and the generation of GI datasets
 Overview of IslandPick pipeline and the generation of GI datasets Predicting GIs using comparative genomics By using whole genome alignments we can identify regions that are present in one genome but not
Overview of IslandPick pipeline and the generation of GI datasets Predicting GIs using comparative genomics By using whole genome alignments we can identify regions that are present in one genome but not
Sequence Analysis '17- lecture 8. Multiple sequence alignment
 Sequence Analysis '17- lecture 8 Multiple sequence alignment Ex5 explanation How many random database search scores have e-values 10? (Answer: 10!) Why? e-value of x = m*p(s x), where m is the database
Sequence Analysis '17- lecture 8 Multiple sequence alignment Ex5 explanation How many random database search scores have e-values 10? (Answer: 10!) Why? e-value of x = m*p(s x), where m is the database
RNA Search and! Motif Discovery" Genome 541! Intro to Computational! Molecular Biology"
 RNA Search and! Motif Discovery" Genome 541! Intro to Computational! Molecular Biology" Day 1" Many biologically interesting roles for RNA" RNA secondary structure prediction" 3 4 Approaches to Structure
RNA Search and! Motif Discovery" Genome 541! Intro to Computational! Molecular Biology" Day 1" Many biologically interesting roles for RNA" RNA secondary structure prediction" 3 4 Approaches to Structure
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Bioinformatics Definitions The use of computational
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Bioinformatics Definitions The use of computational
Bioinformatics Chapter 1. Introduction
 Bioinformatics Chapter 1. Introduction Outline! Biological Data in Digital Symbol Sequences! Genomes Diversity, Size, and Structure! Proteins and Proteomes! On the Information Content of Biological Sequences!
Bioinformatics Chapter 1. Introduction Outline! Biological Data in Digital Symbol Sequences! Genomes Diversity, Size, and Structure! Proteins and Proteomes! On the Information Content of Biological Sequences!
METHODS FOR DETERMINING PHYLOGENY. In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task.
 Chapter 12 (Strikberger) Molecular Phylogenies and Evolution METHODS FOR DETERMINING PHYLOGENY In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task. Modern
Chapter 12 (Strikberger) Molecular Phylogenies and Evolution METHODS FOR DETERMINING PHYLOGENY In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task. Modern
Ch. 9 Multiple Sequence Alignment (MSA)
") Ch. 9 Multiple Sequence Alignment (MSA) - gather seqs. to make MSA - doing MSA with ClustalW - doing MSA with Tcoffee - comparing seqs. that cannot align Introduction - from pairwise alignment to MSA -
Ch. 9 Multiple Sequence Alignment (MSA) - gather seqs. to make MSA - doing MSA with ClustalW - doing MSA with Tcoffee - comparing seqs. that cannot align Introduction - from pairwise alignment to MSA -
CHAPTERS 24-25: Evidence for Evolution and Phylogeny
 CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
Multiple Sequence Alignment, Gunnar Klau, December 9, 2005, 17:
 Multiple Sequence Alignment, Gunnar Klau, December 9, 2005, 17:50 5001 5 Multiple Sequence Alignment The first part of this exposition is based on the following sources, which are recommended reading:
Multiple Sequence Alignment, Gunnar Klau, December 9, 2005, 17:50 5001 5 Multiple Sequence Alignment The first part of this exposition is based on the following sources, which are recommended reading:
Mitochondrial Genome Annotation
 Protein Genes 1,2 1 Institute of Bioinformatics University of Leipzig 2 Department of Bioinformatics Lebanese University TBI Bled 2015 Outline Introduction Mitochondrial DNA Problem Tools Training Annotation
Protein Genes 1,2 1 Institute of Bioinformatics University of Leipzig 2 Department of Bioinformatics Lebanese University TBI Bled 2015 Outline Introduction Mitochondrial DNA Problem Tools Training Annotation
Sequencing alignment Ameer Effat M. Elfarash
 Sequencing alignment Ameer Effat M. Elfarash Dept. of Genetics Fac. of Agriculture, Assiut Univ. amir_effat@yahoo.com Why perform a multiple sequence alignment? MSAs are at the heart of comparative genomics
Sequencing alignment Ameer Effat M. Elfarash Dept. of Genetics Fac. of Agriculture, Assiut Univ. amir_effat@yahoo.com Why perform a multiple sequence alignment? MSAs are at the heart of comparative genomics
Sara C. Madeira. Universidade da Beira Interior. (Thanks to Ana Teresa Freitas, IST for useful resources on this subject)
") Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
The practice of naming and classifying organisms is called taxonomy.
 Chapter 18 Key Idea: Biologists use taxonomic systems to organize their knowledge of organisms. These systems attempt to provide consistent ways to name and categorize organisms. The practice of naming
Chapter 18 Key Idea: Biologists use taxonomic systems to organize their knowledge of organisms. These systems attempt to provide consistent ways to name and categorize organisms. The practice of naming
Chad Burrus April 6, 2010
 Chad Burrus April 6, 2010 1 Background What is UniFrac? Materials and Methods Results Discussion Questions 2 The vast majority of microbes cannot be cultured with current methods Only half (26) out of
Chad Burrus April 6, 2010 1 Background What is UniFrac? Materials and Methods Results Discussion Questions 2 The vast majority of microbes cannot be cultured with current methods Only half (26) out of
Impact of training sets on classification of high-throughput bacterial 16s rrna gene surveys
 (2012) 6, 94 103 & 2012 International Society for Microbial Ecology All rights reserved 1751-7362/12 www.nature.com/ismej ORIGINAL ARTICLE Impact of training sets on classification of high-throughput bacterial
(2012) 6, 94 103 & 2012 International Society for Microbial Ecology All rights reserved 1751-7362/12 www.nature.com/ismej ORIGINAL ARTICLE Impact of training sets on classification of high-throughput bacterial
Model Accuracy Measures
 Model Accuracy Measures Master in Bioinformatics UPF 2017-2018 Eduardo Eyras Computational Genomics Pompeu Fabra University - ICREA Barcelona, Spain Variables What we can measure (attributes) Hypotheses
Model Accuracy Measures Master in Bioinformatics UPF 2017-2018 Eduardo Eyras Computational Genomics Pompeu Fabra University - ICREA Barcelona, Spain Variables What we can measure (attributes) Hypotheses
MULTIPLE SEQUENCE ALIGNMENT FOR CONSTRUCTION OF PHYLOGENETIC TREE
 MULTIPLE SEQUENCE ALIGNMENT FOR CONSTRUCTION OF PHYLOGENETIC TREE Manmeet Kaur 1, Navneet Kaur Bawa 2 1 M-tech research scholar (CSE Dept) ACET, Manawala,Asr 2 Associate Professor (CSE Dept) ACET, Manawala,Asr
MULTIPLE SEQUENCE ALIGNMENT FOR CONSTRUCTION OF PHYLOGENETIC TREE Manmeet Kaur 1, Navneet Kaur Bawa 2 1 M-tech research scholar (CSE Dept) ACET, Manawala,Asr 2 Associate Professor (CSE Dept) ACET, Manawala,Asr
Assessing and Improving Methods Used in Operational Taxonomic Unit-Based Approaches for 16S rrna Gene Sequence Analysis
 APPLIED AND ENVIRONMENTAL MICROBIOLOGY, May 2011, p. 3219 3226 Vol. 77, No. 10 0099-2240/11/$12.00 doi:10.1128/aem.02810-10 Copyright 2011, American Society for Microbiology. All Rights Reserved. Assessing
APPLIED AND ENVIRONMENTAL MICROBIOLOGY, May 2011, p. 3219 3226 Vol. 77, No. 10 0099-2240/11/$12.00 doi:10.1128/aem.02810-10 Copyright 2011, American Society for Microbiology. All Rights Reserved. Assessing
Homology and Information Gathering and Domain Annotation for Proteins
 Homology and Information Gathering and Domain Annotation for Proteins Outline Homology Information Gathering for Proteins Domain Annotation for Proteins Examples and exercises The concept of homology The
Homology and Information Gathering and Domain Annotation for Proteins Outline Homology Information Gathering for Proteins Domain Annotation for Proteins Examples and exercises The concept of homology The
a,bD (modules 1 and 10 are required)
") This form should be used for all taxonomic proposals. Please complete all those modules that are applicable (and then delete the unwanted sections). For guidance, see the notes written in blue and the
This form should be used for all taxonomic proposals. Please complete all those modules that are applicable (and then delete the unwanted sections). For guidance, see the notes written in blue and the
8/23/2014. Phylogeny and the Tree of Life
 Phylogeny and the Tree of Life Chapter 26 Objectives Explain the following characteristics of the Linnaean system of classification: a. binomial nomenclature b. hierarchical classification List the major
Phylogeny and the Tree of Life Chapter 26 Objectives Explain the following characteristics of the Linnaean system of classification: a. binomial nomenclature b. hierarchical classification List the major
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment
 Pairwise sequence alignment") Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Probing diversity in a hidden world: applications of NGS in microbial ecology
 Probing diversity in a hidden world: applications of NGS in microbial ecology Guus Roeselers TNO, Microbiology & Systems Biology Group Symposium on Next Generation Sequencing October 21, 2013 Royal Museum
Probing diversity in a hidden world: applications of NGS in microbial ecology Guus Roeselers TNO, Microbiology & Systems Biology Group Symposium on Next Generation Sequencing October 21, 2013 Royal Museum
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
SnoPatrol: How many snorna genes are there? Supplementary
 SnoPatrol: How many snorna genes are there? Supplementary materials. Paul P. Gardner 1, Alex G. Bateman 1 and Anthony M. Poole 2,3 1 Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton,
SnoPatrol: How many snorna genes are there? Supplementary materials. Paul P. Gardner 1, Alex G. Bateman 1 and Anthony M. Poole 2,3 1 Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton,
The Tree of Life. Chapter 17
 The Tree of Life Chapter 17 1 17.1 Taxonomy The science of naming and classifying organisms 2000 years ago Aristotle Grouped plants and animals Based on structural similarities Greeks and Romans included
The Tree of Life Chapter 17 1 17.1 Taxonomy The science of naming and classifying organisms 2000 years ago Aristotle Grouped plants and animals Based on structural similarities Greeks and Romans included
CHAPTER 10 Taxonomy and Phylogeny of Animals
 CHAPTER 10 Taxonomy and Phylogeny of Animals 10-1 10-2 Copyright The McGraw-Hill Companies, Inc. Permission required for reproduction or display. Linnaeus and Taxonomy More than 1.5 million species of
CHAPTER 10 Taxonomy and Phylogeny of Animals 10-1 10-2 Copyright The McGraw-Hill Companies, Inc. Permission required for reproduction or display. Linnaeus and Taxonomy More than 1.5 million species of
Multiple sequence alignment
 Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
Multiple sequence alignment Multiple sequence alignment: today s goals to define what a multiple sequence alignment is and how it is generated; to describe profile HMMs to introduce databases of multiple
Phylogenetic Analysis. Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center
 Phylogenetic Analysis Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center Outline Basic Concepts Tree Construction Methods Distance-based methods
Phylogenetic Analysis Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center Outline Basic Concepts Tree Construction Methods Distance-based methods
Chapter 17A. Table of Contents. Section 1 Categories of Biological Classification. Section 2 How Biologists Classify Organisms
 Classification of Organisms Table of Contents Section 1 Categories of Biological Classification Section 1 Categories of Biological Classification Classification Section 1 Categories of Biological Classification
Classification of Organisms Table of Contents Section 1 Categories of Biological Classification Section 1 Categories of Biological Classification Classification Section 1 Categories of Biological Classification
Supplementary Information
 Supplementary Information For the article"comparable system-level organization of Archaea and ukaryotes" by J. Podani, Z. N. Oltvai, H. Jeong, B. Tombor, A.-L. Barabási, and. Szathmáry (reference numbers
Supplementary Information For the article"comparable system-level organization of Archaea and ukaryotes" by J. Podani, Z. N. Oltvai, H. Jeong, B. Tombor, A.-L. Barabási, and. Szathmáry (reference numbers